

創薬向けのOSSである「Uni-Mol Docking v2」をローカル環境で動かしてみたいと思います。
実行環境
- OS:WSL2, Ubuntu24.04 (Windows 11 Pro)
- CPU: Intel(R) Core(TM) Ultra 9 185H (WSL2プロセッサ数; 22コア)
- GPU: NVIDIA GeForce RTX 4070 (8GB)
- RAM: 32GB
Uni-Mol Docking V2について
Uni-Mol Docking V2は、2024年5月に公開されたAIベースのドッキングモデルです(Deep Modeling開発)。
AIベースのドッキングモデルとしては2022年に公開されたDiffDockが有名ですが、ベンチマークテストにおいてUni-Mol Docking V2はDiffDockを超える性能を示す有用なモデルです。
今回は、このUni-Mol Docking2 V2について実行環境を構築していきます。また、Protein Data Bank (PDB) に登録されているEGFRの複合体情報を利用し、リドッキング/クロスドッキングによって性能を確認してみたいと思います。
環境構築
Githubを参考に、Ubuntu22.04向けのインストール方法を試します(手順はWSL2環境で成功したものです)。
Uni-Mol Docking V2を動かすためには、①Uni-Coreのインストール、②Uni-Mol Docking V2用のパッケージインストール、③事前学習モデルのダウンロード、④ドッキング用のスクリプト、が必要になります。
今回は専用のconda環境を作成し、必要なパッケージをインストールしていきたいと思います。
まずは、以下のコードでpython3.10をベースとするconda環境を作成し、activateしましょう。
conda create -n unimol-docking2 python=3.10 -y
conda activate unimol-docking2Uni-Coreのインストール
Uni-Coreのインストール方法はいくつかありますが、今回はpipを利用してインストールします。
pipを利用したインストールには、Githubのリポジトリ内のsetup.pyが必要となります。まずはローカル環境にリポジトリをクローンしてリポジトリ内に移動しましょう。クローン先は任意のディレクトリで問題ありません。
cd (任意のディレクトリ)
git clone https://github.com/dptech-corp/Uni-Core.git
cd Uni-Core2025年9月現在、Uni-Coreのインストールには(setup.pyを動かすためには)、モデルを利用するために必要となるPyTorchを事前にインストールする必要があります。
PyTorchのバージョンは最新のもので問題ないようです。公式サイトのインストール方法に従いPyTorchをインストールしておきましょう。
参考までに、今回上手くいったコマンドを記載します。OSはLinux、CUDAのバージョンは12.6を選択しました(CUDAは事前にインストールする必要はありません)。
pip3 install torch torchvision --index-url https://download.pytorch.org/whl/cu126PyTorchのインストールが完了したら、以下のコマンドでUni-Coreをインストールします。
pip install .以上でUni-Coreのインストールは完了です。
Uni-Mol Docking V2用のパッケージインストール
Uni-Coreに加えてRDkitとbiopandasのインストールが必要となります。以下のコードでインストールしましょう。
pip install rdkit-pypi==2022.9.3 -i https://pypi.tuna.tsinghua.edu.cn/simple/ --trusted-host pypi.tuna.tsinghua.edu.cn
pip install biopandas tqdm scikit-learn今回私が環境構築をした際、パッケージのimport時にnumpy関連のバージョンエラーが発生しました。numpyのバージョンを下げることで解決できたので、もし同じ状況に陥った場合は同様の対処をお勧めします(numpy==1.26.4で動作を確認)。
pip install numpy==1.26.4以上で必要なパッケージのインストールは完了です。
事前学習モデルのダウンロード
ドッキング(ポーズ予測)の実行には、訓練済みモデルが必要となります。
Dropboxに訓練済みモデルが公開されているので、こちらをダウンロードしておきましょう。
ドッキング用のスクリプト
これまでの準備で理屈上はドッキングが可能な状態となりました。あとはドッキング用のスクリプトが必要となるだけですが、ゼロベースで用意するのは非常に大変です。
Uni-MolのGithubのリポジトリには、Uni-Mol Docking V2を動かすためのスクリプトが用意されています。最後にリポジトリをローカル環境にクローンしましょう。
cd (任意のディレクトリ)
git clone https://github.com/deepmodeling/Uni-Mol.git以上で環境の準備は完了です。
Uni-Mol Docking V2 の操作
EGFRのリガンド-タンパク質複合体として、5HG8、5UG8、5UG9、5UGC、この4つを利用したリドッキング/クロスドッキングを実施します。
5HG8の結合ポケットを用いて、Uni-Mol Docking V2によるドッキングを行います。
得られたドッキングポーズと各実験構造のリガンド座標に対してRMSD (Root Mean Squared Error) を算出し、ドッキングモデルとしての性能を確認したいと思います。
データ準備
PyMOLを利用して構造の前処理を行います。
詳細は割愛しますが、各構造のリガンドファイル(.sdf)と5HG8のレセプターファイル(.pdb)を用意します。
5HG8のレセプターファイルは、タンパク質の情報のみを残します。結晶水や水分子等の情報はUni-Molの事前学習モデルに考慮されておらず、ノイズになることが考えられるため削除します。
リガンドのインプットファイル作成
※以下、jupyter notebook上での作業を想定しています。
公式リポジトリのnotebookを参考に各種処理を進めます。
まずは、必要となるパッケージのimportとパスを指定を設定します。今後のディレクトリ名やファイル名は適宜読み替えていただくようお願いします。
import json
import numpy as np
import pandas as pd
from tqdm import tqdm
from rdkit import Chem
import os
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem import rdMolAlign
base_dir = '' # PDBから取得した情報と結果を保存するディレクトリを指定
unimol_dir = '' # Uni-Molのリポジトリをクローン下したディレクトリを指定Uni-Mol Docking V2によるドッキングポーズ作成にはリガンドの2D情報が必要です(任意の3D座標をインプットとすることも可能ですが今回は割愛します)。
PDBから取得したリガンドの情報は3Dのため2D情報に直したファイルを準備します。
list_ligs = [
'5HG8', '5UG8', '5UG9', '5UGC'
]
for lig in list_ligs:
sup = Chem.SDMolSupplier(base_dir + f'/data/ligand_{lig}.sdf', removeHs=False)
mol = sup[0]
mol.RemoveAllConformers() # 配座を削除
Chem.MolToMolFile(mol, base_dir + f'/data/input_{lig}.sdf')Gridファイルの作成
配座探索の範囲を指定するためのGridファイルを作成します。
ドッキングシミュレーションでは、結合ポケットの範囲(Grid)を指定して配座探索を行います。今回は5HG8のリガンドの座標から10Åの範囲でGridを指定できるようファイルを作成します。
def calculated_docking_grid_sdf(ligand_path, json_path, add_size=10):
mol = Chem.MolFromMolFile(str(ligand_path), sanitize=False)
coords = mol.GetConformer(0).GetPositions().astype(np.float32)
min_xyz = [min(coord[i] for coord in coords) for i in range(3)]
max_xyz = [max(coord[i] for coord in coords) for i in range(3)]
center = np.mean(coords, axis=0)
size = [abs(max_xyz[i] - min_xyz[i]) for i in range(3)]
center_x, center_y, center_z = center
size_x, size_y, size_z = size
size_x = size_x + add_size
size_y = size_y + add_size
size_z = size_z + add_size
grid_info = {
"center_x": float(center_x),
"center_y": float(center_y),
"center_z": float(center_z),
"size_x": float(size_x),
"size_y": float(size_y),
"size_z": float(size_z)
}
with open(json_path, 'w') as f:
json.dump(grid_info, f, indent=4)
print('Center: ({:.6f}, {:.6f}, {:.6f})'.format(center_x, center_y, center_z))
print('Size: ({:.6f}, {:.6f}, {:.6f})'.format(size_x, size_y, size_z))
ligand_path = base_dir + '/data/ligand_5HG8.sdf'
grid_path = base_dir + f'/data/grid_5HG8_as{add_size}.json'
calculated_docking_grid_sdf(ligand_path, grid_path, add_size=10)メタ情報のファイル作成
利用するリガンドやレセプター、Gridファイルのパスを指定するファイルを作成します。
result_dir = base_dir + '/result'
os.makedirs(result_dir, exist_ok=True)
df = pd.DataFrame(columns=['input_protein', 'input_ligand', 'input_docking_grid', 'output_ligand_name'])
meta_info_file = result_dir + '/test_one2one.csv'
protein_path = base_dir + '/data/receptor_5HG8.pdb'
for i, lig in tqdm(enumerate(list_ligs)):
ligand_path = base_dir + f'/data/input_{lig}.sdf'
predict_name = f'docked_{lig}'
df.loc[i] = [protein_path, ligand_path, grid_path, predict_name]
print(df.info())
print(df.head(3))
df.to_csv(meta_info_file, index= False)ドッキングシミュレーション
jupyterのマジックコマンドを利用してpythonファイルを実行します。
%run $unimol_dir/unimol_docking_v2/interface/demo.py \
--mode batch_one2one \
--batch-size 8 \
--steric-clash-fix \
--conf-size 10 \
--cluster \
--input-batch-file $meta_info_file \
--output-ligand-dir $result_dir \
--model-dir $unimol_dir/unimol_docking_v2_240517.pt引数は参考元から変えていませんが、この条件かつ私の環境では「1化合物あたり10秒弱」でドッキングポーズが算出されました。
RMSDの算出
出てきたドッキングポーズと実験構造の座標を比較します。
def substructure_rmsd(ref_mol, prb_mol, smarts):
ref = Chem.RemoveHs(ref_mol, sanitize=False)
prb = Chem.RemoveHs(prb_mol, sanitize=False)
rms = rdMolAlign.GetBestRMS(prb, ref)
return rms
for lig in list_ligs:
ref_sup = Chem.SDMolSupplier(base_dir + f'/data/ligand_{lig}.sdf', removeHs=False, sanitize=False)
prb_sup = Chem.SDMolSupplier(result_dir + f'/docked_{lig}.sdf', removeHs=False, sanitize=False)
ref_mol = ref_sup[0]
prb_mol = prb_sup[0]
smarts = Chem.MolToSmarts(prb_mol)
rms = substructure_rmsd(ref_mol, prb_mol, smarts)
print(lig, f'RMSD {rms:.3f}Å')出力は、以下の通りです。
5HG8 RMSD 0.545Å
5UG8 RMSD 1.160Å
5UG9 RMSD 0.928Å
5UGC RMSD 1.158Åリドッキング/クロスドッキングは、RMSDが2Å未満であれば良好と言われているため、今回はかなり良い結果なのではないかと思います。
最後にドッキングポーズと実験構造をPyMOLで比べてみましょう。水素が明示的な構造が実験構造です(水素自体はPyMOLで後付け)。
5HG8(RMSD:0.545Å)
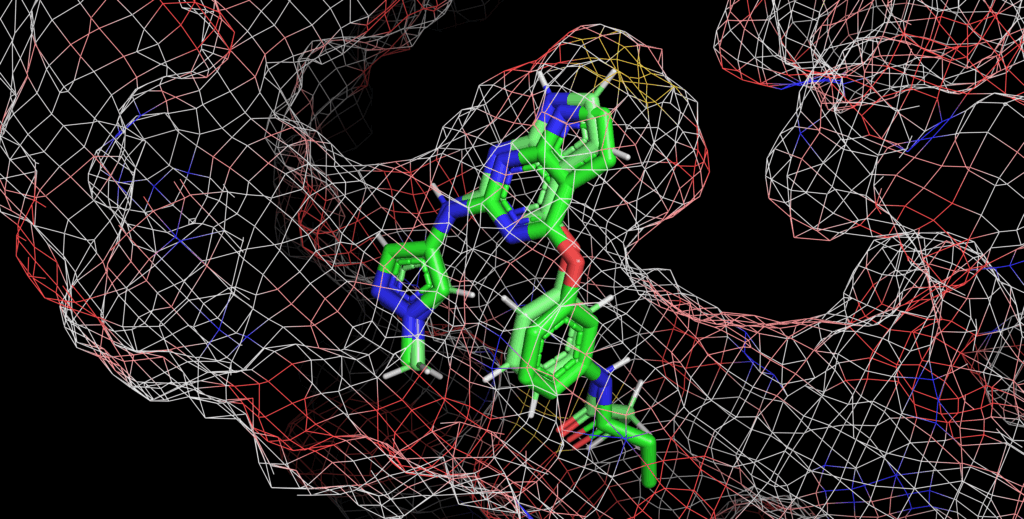
5UG8(RMSD:1.160Å)
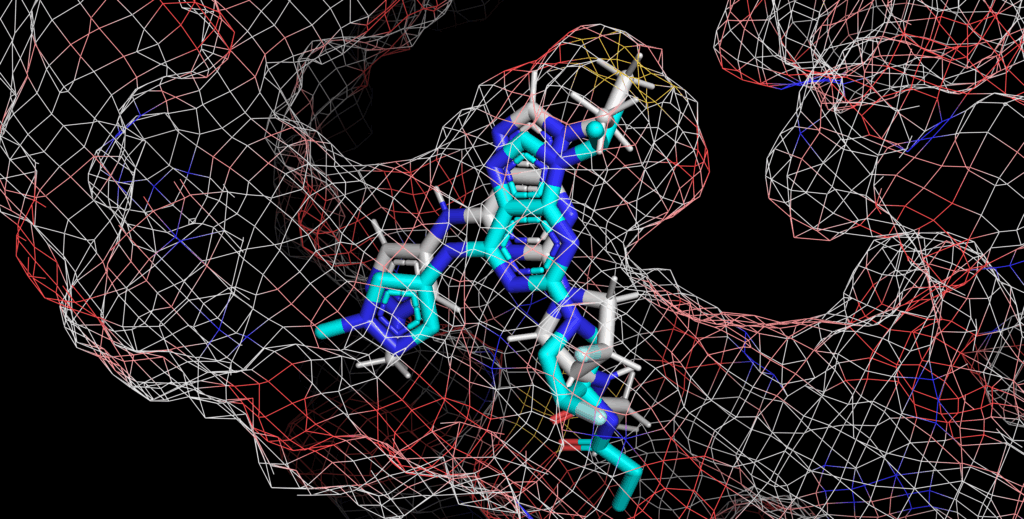
5UG9(RMSD:0.928Å)
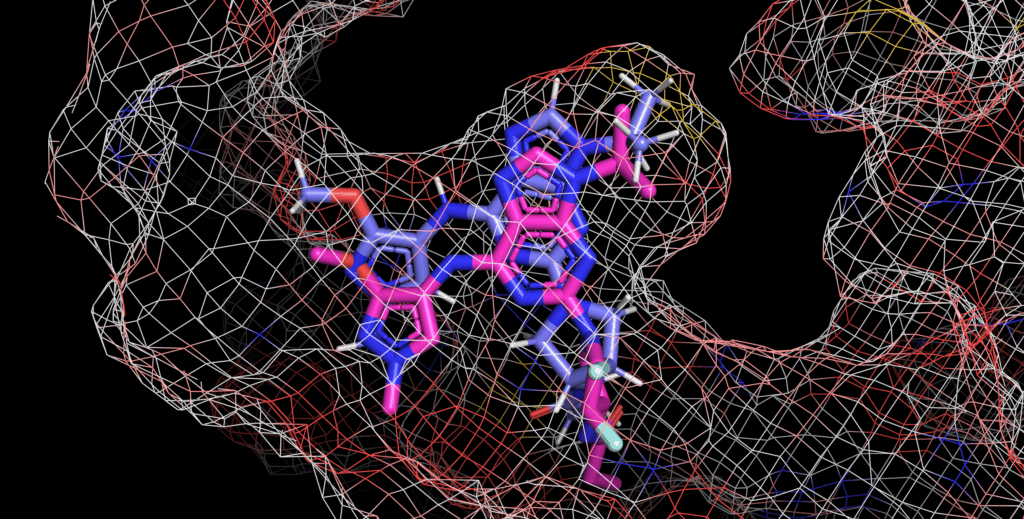
5UGC(RMSD:1.158Å)
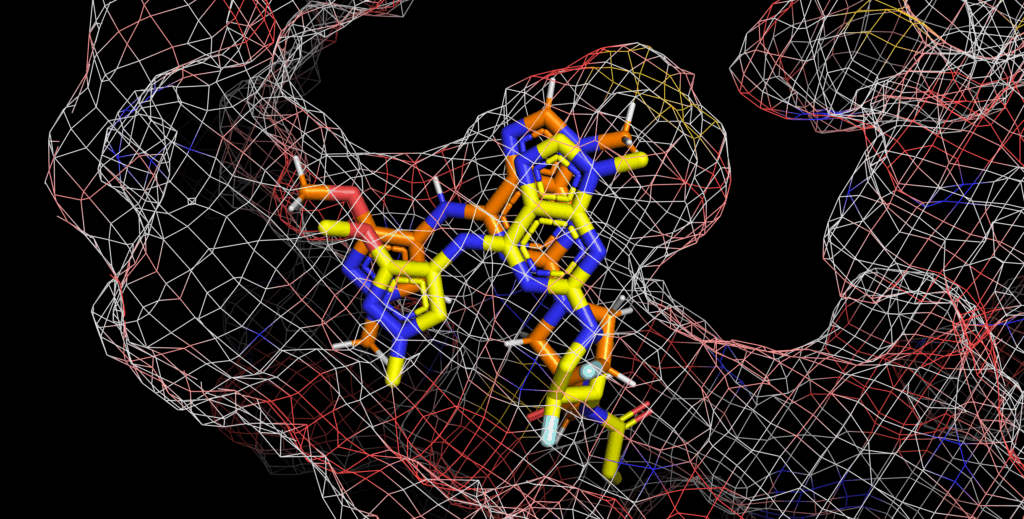
RMSDの通り、実験構造に近いドッキングポーズが算出されています。
構造ベースの印象としては、芳香環に直結しているピロリジン窒素がSp3っぽいこと気になる…くらいでしょうか?
実験構造のようにSp2性の高いほうがエネルギー的に安定だと思います。RMSD上は非常に良い結果ですが、物理化学的な妥当性も考慮されたポーズが出てくるとうれしいですね。エネルギー最適化の後処理で調整可能な範囲だと思いますが、やはりAIベースのドッキングの課題です。
ポーズ生成が早いのでバーチャルスクリーニングの初期段階で利用する分には良いですが、メドケムやSBDDの専門家にこのポーズを持っていくのは少々勇気がいります。
最後に
創薬向けのOSSである「Uni-Mol Docking v2」をローカル環境で動かしてみました。
AIベースのドッキング手法としては、DiffDockなどの手法と並んで有用なツールだと思います。ただ、会社で使うとなると許可が出ないところも多いんですかね?(何故かは言いませんが…日本の会社だと特に)。
最近はSBDD関連で物理化学を考慮したモデルも増えてきているので、引き続き情報を追っていく必要がありそうです。
(2025年9月14日追記)今回利用したコードを記載します。参考になれば幸いです。


コメント