

創薬向けのOSSである「DeepFrag」をローカル環境で動かしてみたいと思います。
実行環境
- OS:WSL2, Ubuntu 24.04 (Windows 11 Pro)
- CPU: Intel(R) Core(TM) Ultra 9 185H (WSL2プロセッサ数; 22コア)
- GPU: NVIDIA GeForce RTX 4070 (8GB)
- RAM: 32GB
DeepFragについて
DeepFragは、2021年に発表されたリード最適化支援の機械学習モデルで、タンパク質結合ポケットの立体情報を取り込みながら、リガンドの置換基変換や分子骨格の伸長に適した候補を提示します。
DeepFragでは、リガンドの官能基を部分的にマスクしたタンパク質–リガンド複合体の結晶構造を入力データとしており、3D-CNNにより抽出された特徴量がマスクされた官能基の分子フィンガープリントに対応するようモデルを最適化しています。
現在、DeepFragを利用するためには以下の3つの方法があります。
- DeepFrag Browser App
- DeepFrag CLI
- DeepFrag API
今回は、簡単に利用できる「DeepFrag Browser App」に加えて、拡張性やソフトウェア連携のような実務運用を見据えて「DeepFrag API」を動かしてみたいと思います。
DeepFrag Browser App
導入
Githubを参考に、WSL2環境でwebアプリサーバーを建ててWindows環境のブラウザから動かしてみます。
手順は以下の通りです。
- Githubのリリースページからzipファイルをダウンロードし、WSL2環境の任意のフォルダに配置
- ファイルを解凍: unzip deepfrag-app.zip
- ディレクトリを移動: cd deepfrag-app
- Webアプリサーバーを起動: python3 -m http.server 8000
- Windows環境のブラウザでWebアプリにアクセス: http://localhost:8000
以下の画像のようなサイトが表示さていれば上手くいっています。
動作確認
DeepFrag Browser Appは、リガンドとタンパク質のファイルをそれぞれ準備することで利用可能です。
今回は、「Use Example Files」ボタンを押すことで利用できるHsPin1pのリガンドついて、ベンゼン環のパラ位に入りうる官能基を探索してみます。
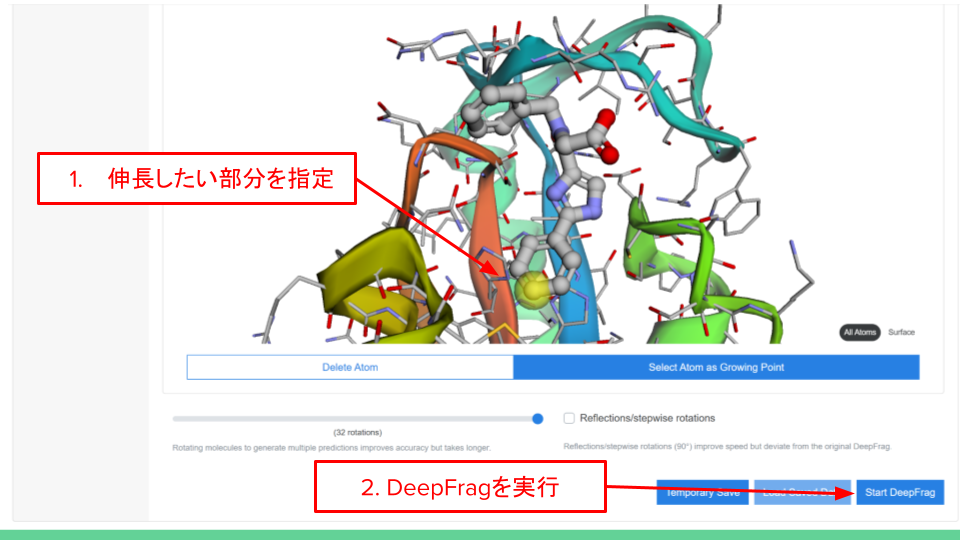
実行後、指定した部分への導入候補の構造とスコアが提示されます。
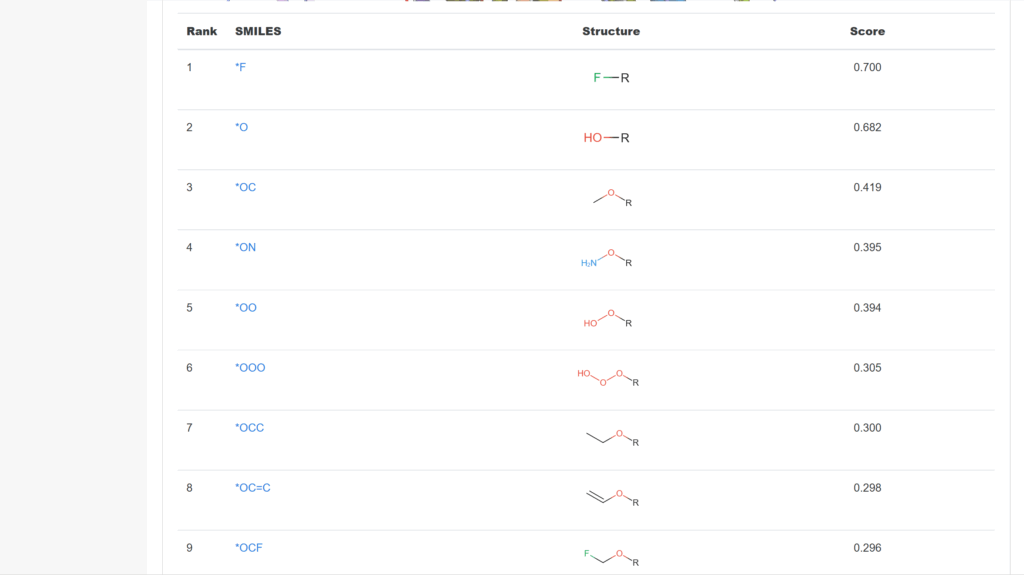
今回のランでは、フッ素が一番目、ヒドロキシ基が二番目に提案されました。
伸長先はフェニルアラニンやメチオニン、ロイシンに囲まれた疎水的な領域っぽいので、フッ素が上位に来るのは妥当なのではないでしょうか?(二番目のヒドロキシ基の妥当性については創薬の経験値が低い私ではわかりかねます…)
複合体の構造情報は必要ですが、非常に簡単な操作で候補が提案されるため、リード最適化段階のアイデア出しに使ってみてもよいかもしれません。
ちなみにDeepFragで提案される置換基は、AIによって生成されたものではなく事前に準備したフラグメントセットから選ばれます。
機械学習モデルのアウトプットは、分子フィンガープリントと同じビット長のベクトルです。このベクトルとフラグメントセットの分子フィンガープリントとのコサイン類似度を算出し、類似度の高い構造が期待度の高い構造として提案されます(Score=コサイン類似度)。
DeepFrag API
DeepFrag Browser Appは簡単に利用できる一方、アプリケーション連携や拡張性を考慮するとAPIで利用するニーズもあるかと思います。
DeepFragの公式リポジトリには、Google Colabで利用できるAPIのサンプルnotebookがあります。今回はこちらを参考に、ローカル環境でAPIの操作確認を実施したいと思います。
導入
ソフトウェア依存を仮想環境で独立して管理したいのでconda環境を作成します。
まずは、任意の保存先へ移動してGitからDeepFragのフォルダをダウンロードします。
cd (任意の保存先)
git clone https://github.com/durrantlab/deepfrag.git次に、DeepFrag用のconda環境を作成します。pythonのバージョンは、Google Colabで利用されている3.10を採用します。パッケージに関しては、Githubからダウンロードしたフォルダ内にあるrequirements.txtに従い、不足分は別途インストールします。
conda create -n deepfrag python=3.10
conda activate deepfrag
cd deepfrag
pip install -r requirements.txt
pip install prody==2.4.1
pip install py3Dmol==2.0.4
pip install torch==2.1.2+cu118 torchvision==0.16.2+cu118 --index-url https://download.pytorch.org/whl/cu118
pip install openbabel-wheel
pip install matplotlib
conda install cudatoolkit=11.8 -y
conda install jupyter -ypip installで失敗する場合は、以下のコマンドを実行しconda環境の作成からお試しください。
sudo apt-get update
sudo apt-get install build-essential python3-dev zlib1g-dev libjpeg-dev libpng-dev
最後に構築済みのモデルとフラグメントセット、テスト用の複合体構造をダウンロードします。
wget -q -O model.zip -L https://durrantlab.pitt.edu/apps/deepfrag/files/final_model_v2.zip
unzip -q model.zip
wget -q -O fingerprints.h5 -L https://durrantlab.pitt.edu/apps/deepfrag/files/fingerprints.h5
wget -L https://files.rcsb.org/download/2XP9.pdb1.gz
gzip -df 2XP9.pdb1.gz
cp ./2XP9.pdb1 ./2XP9.pdb以上でDeepFragの環境構築は完了です。
動作確認
DeepFragのconda環境でnotebookを実行します。流れは基本、Google Colaboの内容に従います。
以下、Jupyter系の環境からの実行を想定してコードを書きます。
ライブラリのインポート
まずは、必要なライブラリをインポートします。
import torch
import prody
import py3Dmol
from rdkit import Chem
from rdkit.Chem import Draw, AllChem
from rdkit.Chem.Draw import IPythonConsole
Draw.IPythonConsole.ipython_3d = True
import numpy as np
import time
import h5py
from tqdm.notebook import tqdm
import matplotlib.pyplot as pltGithubからダウンロードしたフォルダに含まれるコードも利用するため、以下のコードを実行してパスを通しましょう。
import sys
DeepFrag_DIR = "" # Githubからダウンロードしたフォルダの設置場所を入力
sys.path.append(DeepFrag_DIR)
from leadopt.model_conf import LeadoptModel, DIST_FN
from leadopt import grid_util
from leadopt.data_util import REC_TYPER, LIG_TYPER
from leadopt import utilまた、ユーティリティとして2D構造への変換とSDFへの変換用の関数を読み込みます。
def to2d(x):
'''Remove 3d coordinate info from a rdkit mol for display purposes.'''
return Chem.MolFromSmiles(Chem.MolToSmiles(x, isomericSmiles=False))
def tosdf(x):
'''Rdkit mol to SDF string.'''
return Chem.MolToMolBlock(x)+'$$$$\n'構造の前処理
テスト用にPDBからダウンロードした2XP9からリガンドとタンパク質それぞれを取得して保存します。複合体はDeepFrag Browser AppのExampleと同じ複合体です。
# 複合体を読み込む
with open("./2XP9.pdb1", "r") as f:
m = prody.parsePDBStream(f)
# タンパク質とリガンドの指定
rec = m.select('not (nucleic or hetatm) and not water')
lig = m.select('resname 4G8')
# PDBとして保存
prody.writePDB('./rec.pdb', rec)
prody.writePDB('./lig.pdb', lig)
# リガンドはSDFに変換
!obabel lig.pdb -Olig.sdfタンパク質原子の座標と特性を取得
タンパク質原子の座標と特性(原子タイプ、水素結合ドナー/アクセプター、など)を変数に格納します。これらはのちに、3D-CNNへのインプット用のボクセル作成に利用します。
# タンパク質原子の座標と特性を取得
rec_coords, rec_types = util.load_receptor_ob('./rec.pdb')
print('Coords (x,y,z):\n', rec_coords[:5])
print('Types (element, aro, hdon, hacc, charge):\n', rec_types[:5])リガンドの読み込みとフラグメント化
リガンドを読み込んでフラグメントを作成します。伸長の起点となる構造は、ここで作成したフラグメントから選択可能となります。
from rdkit.Chem import rdmolops
# リガンドの読み込み
lig = Chem.MolFromMolFile('./lig.sdf')
ligs = rdmolops.GetMolFrags(lig, asMols = True)
lig = max(ligs, default=lig, key=lambda m: m.GetNumAtoms())
# フラグメント化
frags = util.generate_fragments(lig)
print('Generated %d fragments' % len(frags))
# フラグメントの表示
NUM = 10
Draw.MolsToGridImage(
[to2d(x[0]) for x in frags[:NUM]] + [to2d(x[1]) for x in frags[:NUM]],
legends=(
['Parent %d' % x for x in range(NUM)]
+ ['Fragment %d' % x for x in range(NUM)]
),
molsPerRow=NUM)サンプルでは9番目のフラグメント(ペアレント)を利用して置換基変換を行っています。以下のコードでリガンドの該当部位を確認可能です。
# フラグメントの選択
FRAG_IDX = 9 # 任意のインデックスを選択
# py3Dmolで可視化
view = py3Dmol.view(width=800, height=800)
# タンパク質の設定
view.addModel(open('./rec.pdb', 'r').read(), 'pdb')
view.setStyle({'model': 0}, {'cartoon': {'color':'spectrum'}})
view.addSurface(py3Dmol.VDW,{'opacity':0.7,'color':'white'})
# リガンド(ペアレント)の設定
view.addModel(tosdf(frags[FRAG_IDX][0]), 'sdf')
view.setStyle({'model': 1}, {'stick':{}})
# リガンド(フラグメント)の設定
view.addModel(tosdf(frags[FRAG_IDX][1]), 'sdf')
view.setStyle({'model': 2}, {'stick':{'color': 'yellow'}})
view.zoomTo()リガンド原子の座標と特性を取得
リガンド原子の座標と特性(原子タイプ、水素結合ドナー/アクセプター、など)を変数に格納します。タンパク質原子と同様に、3D-CNNへのインプット用のボクセル作成に利用します。
# リガンド原子の座標と特性を取得
parent = frags[FRAG_IDX][0]
parent_coords = util.get_coords(parent)
parent_types = np.array(util.get_types(parent)).reshape((-1,1))
conn = util.get_connection_point(frags[FRAG_IDX][1])
print('Coords (x,y,z):\n', parent_coords[:5])
print('Types (element):\n', parent_types[:5])
print('Connection point: ', conn)モデルとフラグメントライブラリの読み込み
DeepFragのモデルとフラグメントライブラリを読み込みます。
# CPU計算の設定
USE_CPU = False # 今回はGPUで計算
device = torch.device('cpu') if USE_CPU else torch.device('cuda')
print('Using CPU' if USE_CPU else 'Using GPU')
# モデルの読み込み
model = LeadoptModel.load('./final_model', device=device)
# フラグメントライブラリの読み込み
with h5py.File('./fingerprints.h5', 'r') as f:
f_smiles = f['smiles'][()]
f_fingerprints = f['fingerprints'][()].astype(np.float)
print('Loaded %d fingerprints' % len(f_smiles))ボクセルの設定
モデルへの入力として 3D ボクセルを生成します。DeepFrag では、入力ボクセルのサイズは 24×24×24 です。各ボクセルには 9 つのチャネルがあり、原子の位置や特性に応じて値が割り当てられます。
3D-CNN では、畳み込み演算を通じてボクセル間の情報がやり取りされ、複数のチャネルの値が統合されることで特徴が集約されます。
また、同じ構造であってもインプットの座標が異なると出力が変化します(数学の群論や幾何学の話のようです。私も詳しくは理解できていません)。
ロバストな出力にするために、回転させた入力構造を用意してアウトプットの平均をとります(デフォルトで32)。
start = time.time()
# ボクセルの設定
batch = grid_util.get_raw_batch(
rec_coords, rec_types, parent_coords, parent_types,
rec_typer=REC_TYPER[model._args['rec_typer']],
lig_typer=LIG_TYPER[model._args['lig_typer']],
conn=conn,
num_samples=32,
width=model._args['grid_width'],
res=model._args['grid_res'],
point_radius=model._args['point_radius'],
point_type=model._args['point_type'],
acc_type=model._args['acc_type'],
cpu=USE_CPU
)
batch = torch.as_tensor(batch)
end = time.time()
print('Shape: ', batch.shape)
print('Batch generated in %0.3f seconds' % (end - start))各ボクセルのチャネルの状態は、次のコードで確認可能です(設定はX方向のボクセル24個について)。
batch_n = batch.cpu().numpy()
IDX = 0
SCALE = 0.8
NLAYER = batch_n.shape[1]
NWIDTH = batch_n.shape[2]
plt.figure(figsize=(int(NWIDTH * SCALE), int(NLAYER * SCALE)))
for layer in range(NLAYER):
for x in range(NWIDTH):
ax = plt.subplot(NLAYER, NWIDTH, (layer * NWIDTH) + x + 1)
plt.imshow(batch_n[IDX][layer][x], cmap='jet')
ax.set_xticklabels([])
ax.set_yticklabels([])
plt.subplots_adjust(wspace=0.1, hspace=0.1)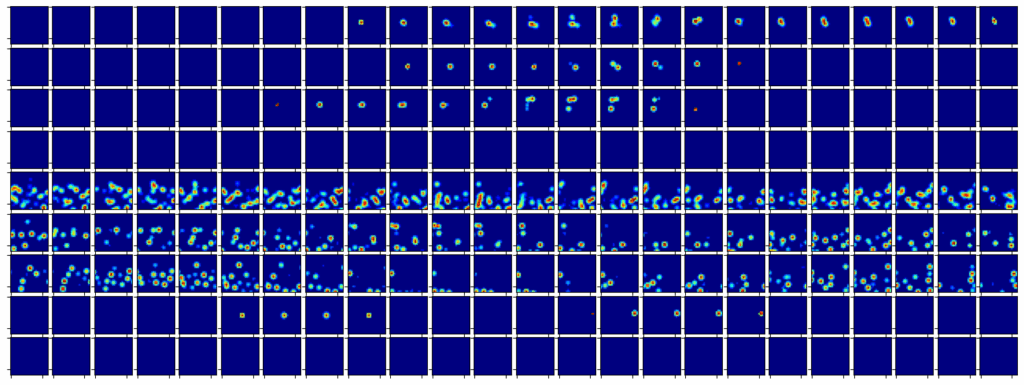
フラグメントの予測
フラグメントの予測を実行します。予測は数秒で完了します。
start = time.time()
pred = model.predict(batch.float()).cpu().numpy()
end = time.time()
print('Generated prediction in %0.3f seconds' % (end - start))
print('Shape:', pred.shape)アウトプットは、2,048次元のベクトル×32です。各ベクトルの凡その値は以下のコードで確認できます(各次元は0~1)。
IDX = 0
fp = pred[IDX].reshape((32, 64))
plt.imshow(fp)
フラグメントライブラリとの比較
アウトプットのベクトルとフラグメントライブラリの類似度を計算し、類似度上位16構造を表示します。
# フィンガープリントを平均化
avg_fp = np.mean(pred, axis=0)
print('Average FP shape:', avg_fp.shape)
# 関数の設定
dist_fn = DIST_FN[model._args['dist_fn']]
# コサイン類似度(?)を計算
dist = dist_fn(
torch.Tensor(avg_fp).unsqueeze(0),
torch.Tensor(f_fingerprints))
# SMILESと類似度の照合
dist = list(dist.numpy())
scores = list(zip(f_smiles, dist))
scores = sorted(scores, key=lambda x:x[1])
# トップ16のフラグメントを表示
print('Top 16 fragments:')
mols = [Chem.MolFromSmiles(x[0]) for x in scores[:16]]
leg = ['Dist: %0.3f' % x[1] for x in scores[:16]]
Draw.MolsToGridImage(mols, molsPerRow=4, legends=leg)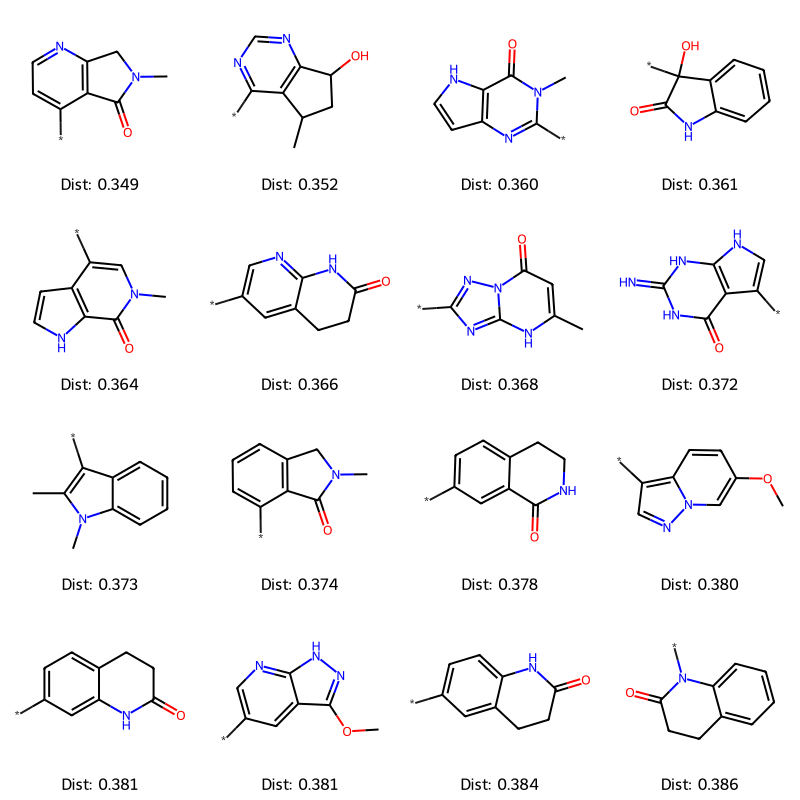
Distはコサイン類似度から算出されている値のはずですが、高ければよいのか低ければよいのか正直よくわかりません。1-類似度で低いほうが似ていると考えるのでしょうか?
ペアレントとフラグメントの結合
ペアレントとフラグメントを結合し、完全な構造を取得します。
以下の関数は、3D空間での結合と力場レベルでのエネルギー安定化(二面角スキャン)を実施しているようです(理解が及んでいない部分がありますので元コードそのまま)。
def geometric_embedding(fragment):
"""Generate a realistic 3D embedding of a fragment.
Note: this method strictly optimizes the fragment conformation without respect
to the receptor or parent molecule. For small fragments, the conformation will
already be fairly fixed so this probably won't do much.
Args:
- fragment: rdkit molecule
Returns:
- an rdkit molecule with 3D coordinates
"""
frag = to2d(fragment)
# Temporarily replace dummy atoms with hydrogen so we get reasonable geometry.
dummy_idx = [x.GetIdx() for x in fragment.GetAtoms() if x.GetAtomicNum() == 0]
for idx in dummy_idx:
frag.GetAtomWithIdx(idx).SetAtomicNum(1)
# Minimize engergy.
frag = Chem.AddHs(frag)
cids = AllChem.EmbedMultipleConfs(frag, 50, pruneRmsThresh=1)
for conf in cids:
AllChem.UFFOptimizeMolecule(frag, confId=conf, maxIters=200)
# Replace any dummy atoms.
for idx in dummy_idx:
frag.GetAtomWithIdx(idx).SetAtomicNum(0)
return frag, cids
def get_connecting_atoms(mol):
"""Return the connection point atom (element 0), it's neighbor,
and a neighbor of the neighbor (for defining dihedral)."""
connectidx = [a.GetIdx() for a in mol.GetAtoms() if a.GetAtomicNum() == 0][0]
atm = mol.GetAtomWithIdx(connectidx)
nextatm = atm.GetNeighbors()[0]
nextidx = nextatm.GetIdx()
nextnextidx = [a for a in nextatm.GetNeighbors() if a.GetIdx() != connectidx][0].GetIdx()
return connectidx, nextidx, nextnextidx
def embed_fragment(rec, parent, fragment):
"""Generate an embedding of the (parent/fragment) fusion molecule in the
context of a receptor.
Args:
- rec: An rdkit mol containing the receptor complex.
- parent: An rdkit mol containing the parent molecule.
- fragment: An rdkit mol containing the fragment molecule that should be
attached to the parent.
Returns:
(ligand, energies, best_energy)
- ligand: The optimized parent/fragment ligand as an rdkit mol.
- energies: A list of predicted energies from sampling different conformations.
- best_energy: The lowest energy from the sampled conformations.
"""
energies = []
Chem.SanitizeMol(rec)
fragment, cids = geometric_embedding(fragment)
# Find the dihedral
paridx, parnext, parnextnext = get_connecting_atoms(parent)
best_energy = np.inf
best_mol = None
# For each conformer...
for cid in tqdm(cids, desc='Sampling conformations'):
mol = Chem.RWMol(fragment, False, cid)
# Align the connection point.
fragidx, fragnext, fragnextnext = get_connecting_atoms(mol)
Chem.rdMolAlign.AlignMol(
mol, parent, atomMap=[(fragidx,parnext),(fragnext,paridx)])
# Merge into new molecule.
merged = Chem.RWMol(Chem.CombineMols(parent,mol))
# Update fragment indices.
fragidx += parent.GetNumAtoms()
fragnext += parent.GetNumAtoms()
fragnextnext += parent.GetNumAtoms()
bond = merged.AddBond(parnext,fragnext,Chem.rdchem.BondType.SINGLE)
merged.RemoveAtom(fragidx)
merged.RemoveAtom(paridx)
Chem.SanitizeMol(merged)
# Update indices to account for deleted atoms.
if fragnext > fragidx: fragnext -= 1
if fragnextnext > fragidx: fragnextnext -= 1
fragnext -= 1
fragnextnext -= 1
if parnext > paridx: parnext -= 1
if parnextnext > paridx: parnextnext -= 1
# Optimize the connection of the fragment (bond is wrong length).
ff = AllChem.UFFGetMoleculeForceField(merged)
for p in range(parent.GetNumAtoms()-1): # Don't include dummy atom.
ff.AddFixedPoint(p) # Don't move parent.
ff.Minimize()
# Create a complex with the receptor.
reclig = Chem.CombineMols(rec, merged)
Chem.SanitizeMol(reclig)
# Determine dihedral indices.
l = fragnextnext+rec.GetNumAtoms()
k = fragnext+rec.GetNumAtoms()
j = parnext+rec.GetNumAtoms()
i = parnextnext+rec.GetNumAtoms()
# Sample the dihedral.
for deg in tqdm(range(0,360,5), desc='Sampling dihedral angle'):
Chem.rdMolTransforms.SetDihedralDeg(reclig.GetConformer(),i,j,k,l,deg)
# Create forcefield for the whole complex.
# reclig = Chem.AddHs(reclig)
ff = AllChem.UFFGetMoleculeForceField(reclig,ignoreInterfragInteractions=False)
# Fix everything but the fragment.
for p in range(rec.GetNumAtoms()+parent.GetNumAtoms()-1):
ff.AddFixedPoint(p)
energy = ff.CalcEnergy()
energies.append(energy)
if energy < best_energy:
best_energy = energy
best_mol = Chem.RWMol(reclig)
# Extract the best ligand.
ligatoms = set(range(rec.GetNumAtoms(), best_mol.GetNumAtoms()))
ligbonds = [b.GetIdx() for b in best_mol.GetBonds() if b.GetBeginAtomIdx() in ligatoms and b.GetEndAtomIdx() in ligatoms]
bestlig = Chem.PathToSubmol(best_mol, ligbonds)
return bestlig, energies, best_energy類似度がトップのフラグメントの中から完全な構造を取得するフラグメントを選びます。こちらも元コードそのままですが、トップスコアのフラグメントを選んでいるようです。また、最後の可視化用にタンパクの構造も読み込んでいます。
rec = Chem.MolFromPDBFile('rec.pdb') # Load receptor into rdkit.
fragment = mols[0] # Pick the top-predicted fragment.
Draw.MolsToGridImage(
[to2d(parent), to2d(fragment)],
legends=['Parent', 'Fragment'])関数を適用して構造を取得します。プロットも表示されますが二面角スキャンのプロットになります。
lig, energies, best_energy = embed_fragment(rec, parent, fragment)
plt.plot(energies)
plt.ylim(0,10000)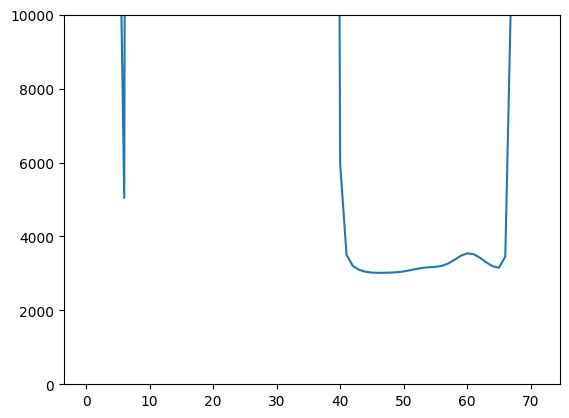
最後に元の構造とDeepFragで提案された構造をpy3Dmolで可視化してみます。黄色が元構造です。
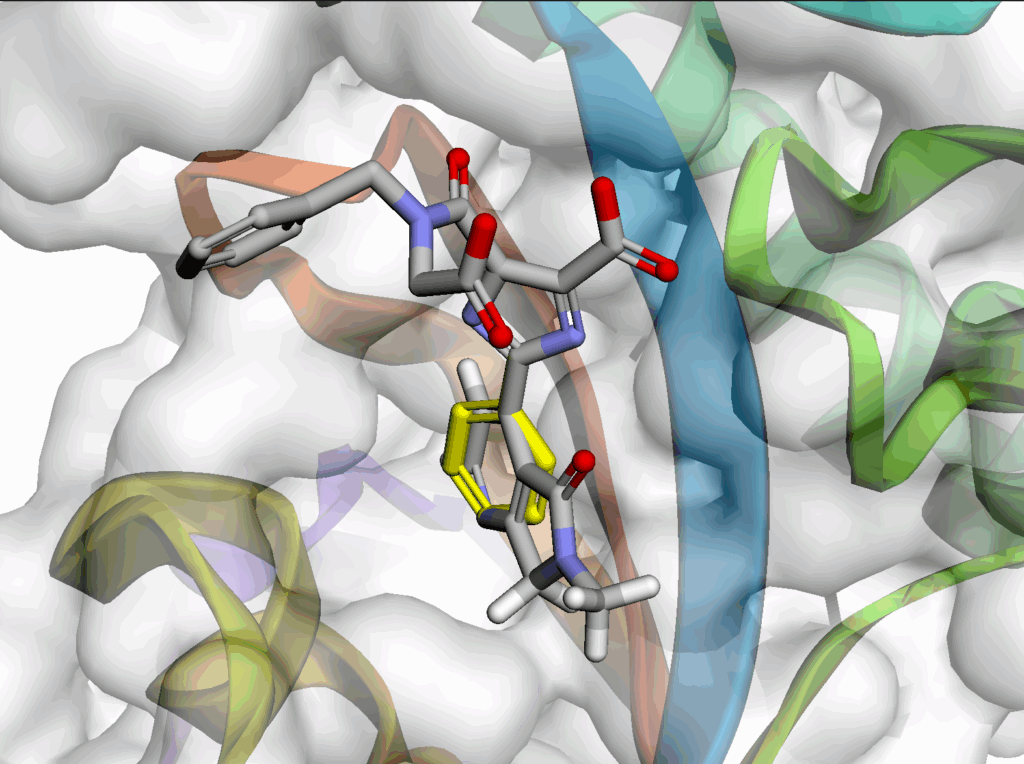
…いい構造なのかな?私の経験値だと見ただけではよくわかりません。
また、提案された構造の3D座標は結合ポケットを考慮していないので、改めてドッキングシミュレーションしてみる必要があるかもしれません。
今回は保存しませんが、変数ligは提案構造のMolオブジェクト(RDkit)なのでSDFへの保存も容易にできます。
最後に
創薬向けのOSSである「DeepFrag」をローカル環境で動かしてみました。
DeepFrag Browser Appは、導入も操作も簡単かつ構造の提案スピードを速いので、入りうる官能基の候補に見落としがないか確認する分には使い勝手が良いと感じました。
DeepFrag APIは、一部理解が及ばなかった部分があるため、自分が使いこなすにはもう少し勉強が必要そうです。
データドリブンな化合物提案がなされているため、ターゲットによっては非常に有効に機能するのではないかという期待があります。APIを使いこなしてワークフロー化できると幅が広がりそうですね。


コメント