
オープンソースソフトウェアを用いて「Grid Inhomogeneous Solvation Theory (GIST)」による水解析を実施します。※今回利用したコードはこちら
実行環境
- OS: Ubuntu 22.04.3 LTS
- CPU: AMD Ryzen 7 5700X 8-Core Processor
- GPU: NVIDIA GeForce RTX 3060 Ti (8G)
- RAM: 32GB
GIST について
GISTとは、MDシミュレーション中の水分子の振る舞いを空間グリッド上で統計力学的に解析する手法です。
直感的には、「このポケットに存在する水は居心地が良いのか、それとも悪いのか?」を、エネルギー項とエントロピー項に分解して定量化する方法と捉えることができます。
創薬では、
- この水はどかしたほうがいい?
- この水は残したほうがいい?
- どかすならはどんな置換基を入れれば得か?
といった問いを常に考えながら化合物設計を行います。
GISTは、これらの判断に対して物理的な根拠を与えてくれる解析手法です。
本記事では、OpenMM と AmberTools を用いて GIST 解析を実施し、タンパク質ポケット中の水の安定性を評価する手順を紹介します。
環境構築
※NVIDIAのGPU環境を想定しています。NVIDIAのGPUがない、あるいはGPUドライバーをインストールしていない環境では、適切な動作となりませんのでご注意ください。
以前に、OpenMMを用いたMDシミュレーションの実施方法を取り扱いました。その際と同様のconda環境を利用します。環境構築の方法は以下の記事をご参考ください。

また、GIST解析用の補助ツールとしてGISTPPを利用します。以下の手順でコンパイルしておきましょう。
cd (任意のディレクトリ)
git clone https://github.com/gosldorf/gist-post-processing.git
cd gist-post-processing
g++ -o gistpp gistpp.cpp gistpp_main.cppGIST解析
構築したconda環境からMDシミュレーションを実行します。コードの実行は”Jupyter Notebook”、分子の可視化は”PyMOL”を想定しています。
ファイル準備
ファイルの前処理に必要となるPythonパッケージをインポート、および作業ディレクトリのパスを変数として設定します。
import os, requests
from collections import defaultdict
from Bio import PDB
from pdbfixer import PDBFixer
from openmm.app import PDBFile
base_dir = "" # 任意のディレクトリ次に、今回利用する「血液凝固因子Xa(factor X)」のX線構造(PDB:1FJS)をダウンロードし、MDシミュレーションに向けて、レセプターのファイルとリガンドのファイルに分割します。
リガンドのファイルは結合情報がうまく読み取れないため、Python API/コマンドラインからの作成が困難です。1FJSのPDBファイルをPyMOLで読み込み、リガンドだけSDFとして保存しましょう(作成方法割愛)。
# ファイルのダウンロード(1FJS: Factor Xa)
pdb_id = "1FJS"
pdb_id = pdb_id.lower()
url = f"https://files.rcsb.org/download/{pdb_id}.pdb"
os.makedirs(base_dir + "/data", exist_ok=True)
out_file = base_dir + f"/data/{pdb_id}.pdb"
response = requests.get(url)
if response.status_code == 200:
with open(out_file, "wb") as f:
f.write(response.content)
else:
print(f"Failed to fetch {pdb_id} from OPM (status {response.status_code})")
# リガンドとレセプターの分割
protein_file = base_dir + f"/data/receptor_{pdb_id}.pdb"
ligand_sdf = base_dir + f"/data/ligand_{pdb_id}.sdf" # PyMOLで作成
ligand_resname = "Z34"
parser = PDB.PDBParser(QUIET=True)
structure = parser.get_structure("complex", out_file)
io = PDB.PDBIO()
# レセプターを抽出(チェインごとに「残す」残基範囲を指定)
class ProteinSelect(PDB.Select):
def __init__(self, chain_include_ranges: dict[str, list[tuple[int, int]]]):
self.chain_include_ranges = chain_include_ranges
def accept_residue(self, residue):
# 標準アミノ酸のみ
if not PDB.is_aa(residue, standard=True):
return False
chain_id = residue.get_parent().id # チェインID
res_id = residue.id[1] # residue number
# 指定チェインでなければ除外
if chain_id not in self.chain_include_ranges:
return False
# 指定された範囲に入っていれば残す
for lo, hi in self.chain_include_ranges[chain_id]:
if lo <= res_id <= hi:
return True
return False
include = {
"A": [(16, 244)],
"L": [(87, 52)],
}
io.set_structure(structure)
io.save(protein_file, ProteinSelect(include))1FJSには、altLocで登録されているアミノ酸残基が複数存在します。今回は厳密性を求めていないため、一番最初に登録されている残基を選択しシミュレーションを実施します。
# レセプターファイルの処理
protein_file2 = base_dir + f"/data/receptor_{pdb_id}_del.pdb"
def strip_icode_auto_lines(pdb_lines):
has_blank = defaultdict(bool)
# 1st pass: 無印が存在する残基を記録
for l in pdb_lines:
if l.startswith(("ATOM ", "HETATM")) and len(l) >= 27:
key = (l[21], l[22:26].strip()) # (chain, resSeq)
if l[26] == " ":
has_blank[key] = True
# 2nd pass: 無印がある残基では iCode 付き行を除去
out = []
for l in pdb_lines:
if l.startswith(("ATOM ", "HETATM")) and len(l) >= 27:
key = (l[21], l[22:26].strip())
if has_blank.get(key, False) and l[26] != " ":
continue
out.append(l)
return out
with open(protein_file, mode="r") as f:
lines = f.readlines()
cleaned = strip_icode_auto_lines(lines)
with open(protein_file2, mode="w") as f:
f.writelines(cleaned)
# PDBfixer
protein_file_fixed = base_dir + f"/data/receptor_{pdb_id}-fixed.pdb"
fixer = PDBFixer(filename=protein_file2)
fixer.findNonstandardResidues()
fixer.replaceNonstandardResidues()
fixer.removeHeterogens(keepWater=False)
fixer.findMissingResidues()
fixer.findMissingAtoms()
fixer.addMissingAtoms()
with open(protein_file_fixed, "w") as f:
PDBFile.writeFile(fixer.topology, fixer.positions, f, keepIds=True)レセプターのファイルは、AmberToolsでの操作用に追加で前処理を行います。
# Amber用にファイルを変換
protein_file_amber = base_dir + f"/data/receptor_{pdb_id}_amber.pdb"
%cd {base_dir}/data
%mkdir temp
%cd temp
!pdb4amber -i {protein_file_fixed} -o {protein_file_amber} --nohyd
%cd {base_dir}/data
!rm -r {base_dir}/data/temp系の準備
シミュレーションに必要となる情報や水分子やイオンを含んだ系を準備します。
MDシミュレーションはOpenMMで実施しますが、過去記事で紹介した手順(OpenMM関連のパッケージで力場パラメータを作成する方法)ではGIST解析用のパラメータファイルが不足します。
OpenMM関連のパッケージで補完する方法はあるかもしれませんが、今回は安全策を取ってAberToolsで系を作成します。
まずは利用する各種ライブラリをインポートしましょう。
import subprocess, textwrap次に、antechamberとparmchk2を用いてリガンドのパラメータファイルを作成します。
# リガンドのパラメータを作成
ligand_frcmod = base_dir + f"/data/ligand_{pdb_id}.frcmod"
ligand_mol2 = base_dir + f"/data/ligand_{pdb_id}.mol2"
%cd {base_dir}/data
%mkdir temp
%cd temp
!antechamber -i {ligand_sdf} -fi sdf -o {ligand_mol2} -fo mol2 -c bcc -nc 1 -at gaff2
!parmchk2 -i {ligand_mol2} -f mol2 -o {ligand_frcmod}
%cd {base_dir}/data
!rm -r {base_dir}/data/tempレセプターのパラメータ化や水・イオンの追加、リガンドの統合などは、tleapを利用します。実行のために以下のコードで”tleap.in”を作成します。
# tleap.inを作成
parm7_file = base_dir + f"/data/system_{pdb_id}.parm7"
rst7_file = base_dir + f"/data/system_{pdb_id}.rst7"
system_file = base_dir + f"/data/system_{pdb_id}.pdb"
leap_in_path = base_dir + "/data/tleap.in"
tleap_in = f"""
source leaprc.protein.ff14SB
source leaprc.water.tip3p
source leaprc.gaff2
# ligand パラメータ
loadamberparams {ligand_frcmod}
LIG = loadmol2 {ligand_mol2}
# protein
PROT = loadpdb {protein_file_amber}
# 複合体
COMPLEX = combine {{ PROT LIG }}
# 溶媒化
solvatebox COMPLEX TIP3PBOX 10.0
# イオン追加
addions COMPLEX Na+ 0
addions COMPLEX Cl- 0
# 出力
saveamberparm COMPLEX {parm7_file} {rst7_file}
savepdb COMPLEX {system_file}
quit
"""
with open(leap_in_path, mode="w") as f:
f.write(textwrap.dedent(tleap_in))tleap.inが作成できたら、以下のコードでtleapを実行します。
# tleap実行(ログ保存)
leap_log_path = base_dir + "/data/leap.log"
cmd = ["tleap", "-f", leap_in_path]
res = subprocess.run(cmd, capture_output=True, text=True)
with open(leap_log_path, "w") as f:
f.write(res.stdout)
f.write("\n\n[STDERR]\n")
f.write(res.stderr)
print("tleap return code:", res.returncode)
print("leap.in:", leap_in_path)
print("leap.log:", leap_log_path)
print("parm7 exists:", os.path.exists(parm7_file))
print("rst7 exists:", os.path.exists(rst7_file))
# 失敗時はログのERROR行だけ抜く
if res.returncode != 0 or (not os.path.exists(parm7_file)) or (not os.path.exists(rst7_file)):
print("\n---- tleap errors (grep) ----")
for line in (res.stdout + "\n" + res.stderr).splitlines():
if "ERROR" in line.upper() or "FATAL" in line.upper() or "Could not" in line:
print(line)
raise RuntimeError("tleap failed; check leap.log")MDシミュレーション
以下のステップでMDシミュレーションを実行します。
- エネルギー最適化
- 等温緩和
- 昇温
- 平衡化
- 生産
MDシミュレーションは以前に記事でも取り扱ったため解説を割愛します。今回の詳細なコードは、Githubをご確認ください。
GIST
利用するパッケージのインポート、及び作業ディレクトリのパス設定・作成等を行います。
import copy
import pytraj as pt
gist_dir = base_dir + "/GIST"
os.makedirs(gist_dir, exist_ok=True)
gistpp_dir = "/home/shaeo/opt/gist-post-processing"解析するグリッドを指定するために、リガンドの中心座標を取得します。
”nc_path_md”には、生産MDのトラジェクトリ(.nc、後処理実施済み)を指定しています。こちらの変数は、MDシミュレーションのコードを実行することで作成されます。
# リガンドの中心座標を算出
traj = pt.load(nc_path_md, parm7_file)
mask = traj.top.select(':MOL')
coords = traj.xyz[0][mask]
center = coords.mean(axis=0)
X, Y, Z = center
print(X, Y, Z)以下のコードでGIST解析を実行します。
# gist.inを作成
gist_in_file = gist_dir + "/gist.in"
gist_in = f"""
parm {parm7_file}
trajin {nc_path_md}
gist gridcntr {X:.6f} {Y:.6f} {Z:.6f} griddim 60 60 60 gridspacn 0.5 temp 300.0 out gist
go
"""
with open(gist_in_file, mode="w") as f:
f.write(textwrap.dedent(gist_in))
# GIST
%cd {gist_dir}
!cpptraj -i {gist_in_file}
%cd {base_dir}これで結合ポケット周辺における水解析の結果ファイル(.dx)が作成されました。
それぞれのファイルには、各グリッドにおけるエネルギーや密度の情報が含まれています。重要なものについては、以下の通りです(参考:https://ambermd.org/tutorials/advanced/tutorial25/section3.php)
- gist-Esw-dens.dx:密度加重溶質-水相互作用エネルギー (kcal/mol/Å3)
- gist-Eww-dens.dx: 密度加重水-水相互作用エネルギー(kcal/mol/Å3)
- gist-gO.dx:ボクセル内で見つかった酸素中心の数密度(バルク水密度との比率)
- gist-dTStrans-dens.dx:密度加重一次並進エントロピー (kcal/mol/Å3)
- gist-dTSorient-dens.dx :密度重み付け一次配向エントロピー (kcal/mol/Å3)
最後に、上記のファイルを組み合わせて溶媒自由エネルギーの結果ファイルを作成します。
# 総溶媒エネルギーを算出
dx_Esw_dense = gist_dir + "/gist-Esw-dens.dx"
dx_hald_Esw_dense = gist_dir + "/half-gist-Esw-dens.dx"
dx_Eww_dense = gist_dir + "/gist-Eww-dens.dx"
dx_Etot_dense = gist_dir + "/gist-Etot-dens.dx"
dx_Etot = gist_dir + "/gist-Etot.dx"
%cd {gistpp_dir}
!./gistpp -i {dx_Esw_dense} -op multconst -opt const 0.5 -o {dx_hald_Esw_dense}
!./gistpp -i {dx_Eww_dense} -i2 {dx_hald_Esw_dense} -op add -o {dx_Etot_dense}
!./gistpp -i {dx_Etot_dense} -op multconst -opt const 0.125 -o {dx_Etot}
# エントロピーを集約
dx_dTStrans_dense = gist_dir + "/gist-dTStrans-dens.dx"
dx_dTSorient_dense = gist_dir + "/gist-dTSorient-dens.dx"
dx_dTStot_dense = gist_dir + "/gist-dTStot_dense.dx"
dx_dTStot = gist_dir + "/gist-dTStot.dx"
!./gistpp -i {dx_dTStrans_dense} -i2 {dx_dTSorient_dense} -op add -o {dx_dTStot_dense}
!./gistpp -i {dx_dTStot_dense} -op multconst -opt const 0.125 -o {dx_dTStot}
# 溶媒自由エネルギーを算出
dx_dA = gist_dir + "/gist-dA.dx"
!./gistpp -i {dx_Etot} -i2 {dx_dTStot} -op sub -o {dx_dA}
%cd {base_dir}可視化(PyMOL)
溶媒自由エネルギーの結果ファイルから、高溶媒密度と高溶媒エネルギーを含む水和サイトを可視化します。
PyMOL上で、1. MDシミュレーションのエネルギー最適化後のPDBファイル、2. 溶媒自由エネルギーの結果ファイルを読み込ませたあと、以下のコマンドを実行します。
isosurface dA_good, gist-dA, -0.5
color cyan, dA_good
set transparency, 0.5, dA_good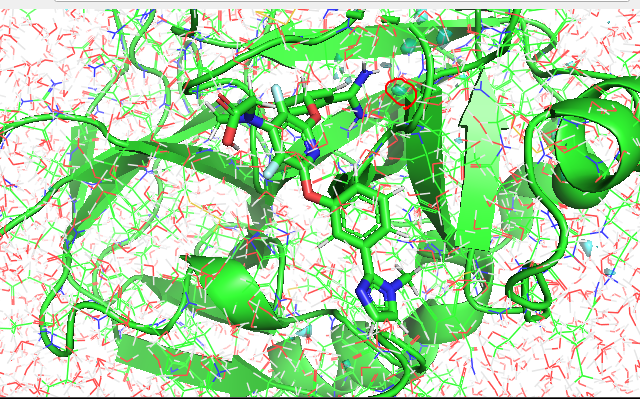
すごく見づらいですが、水色の部分が不安定な水和サイトとなります。
赤枠で囲んだサイトはポケットの比較的深い部分に位置しています。実務で活用する際は、この水が構造水か置換可能な水かを見極めることが重要となります。
最後に
オープンソースソフトウェアを利用してGISTによる水解析を実施しました。
創薬を進める上では非常に有用な解析手法だと思う一方、実務での活用のためには「水の影響を議論できる文化の醸成」が障壁になると感じました。
in silicoの中でも比較的理解に時間を要する分野なので、少しづつでも周りの目に留まるように解析・共有を続けていくが必要がありそうです。


コメント