

「OpenMM」を利用して膜タンパク質+リガンドの分子動力学(MD)シミュレーションを行います。
実行環境
- OS: Ubuntu 22.04.3 LTS
- CPU: AMD Ryzen 7 5700X 8-Core Processor
- GPU: NVIDIA GeForce RTX 3060 Ti (8G)
- メモリ: 32GB
OpenMM について
OpenMMは、GPUアーキテクチャに最適化されたMDエンジンであり、柔軟なPython APIとC++コアを備えたオープンソースプラットフォームです。
Python から数行でシミュレーションを記述できる柔軟さと、Amber・CHARMM・GROMACS など主要フォーマットを直接扱える互換性を備えています。
「自分でプロトコルを書き換えたい」「GPU を最大限に使いたい」といったニーズに応える設計になっており、研究者が自由に拡張できるのが大きな魅力です。
今回は、OpenMMを利用して、膜タンパク質とリガンドのMDシミュレーションを行います。
環境構築
※NVIDIAのGPU環境を想定しています。NVIDIAのGPUがない、あるいはGPUドライバーをインストールしていない環境では、適切な動作となりませんのでご注意ください。
公式サイト、「TeachOpenCADD」、「making-it-rains」を参考にconda環境を構築します。
以下、私の環境で実際に利用したコマンドです。
conda install -n base mamba # mambaをインストールしていない場合
conda create -n openmm python=3.10
conda activate openmm
mamba install -y openmm cuda-version=12.6 # nividia-smiの出力に応じて変更
mamba install -y mdtraj openmmforcefields openff-toolkit pdbfixer pypdb rdkit jupyter biopython openbabel pymol-open-source最新バージョンのOpenMMは、CUDAランタイムが同時にインストールされるため、自身でcuda-toolkitをインストールすることなくGPU計算が実行可能です。
ただし、デフォルトでインストールされるCUDAのバージョンは比較的新しいため、環境によってはGPUとバージョンが合わない可能性があります(私の環境でもデフォルトは動きませんでした)。
4行目の”cuda-version”でバーション指定が可能なため、計算がうまく行かない場合は自身の環境に適したバージョンに変更しましょう。
膜タンパク質+リガンドのMDシミュレーション
構築したconda環境からMDシミュレーションを実行します。コードの実行は”Jupyter Notebook”、分子の可視化は”PyMOL”を想定しています。
また、私はあまりMDに詳しい人間ではありません。論文等も参考にしておりますが、条件設定に関しては参考程度に見ていただければと思います。
ファイル準備
膜タンパク質として、「orexin receptor 1 (OX1)」のX線構造(PDB:4ZJC)を利用します。
4ZJCの構造情報はPDBではなく、OPM (Orientations of Proteins in Membranes) のデータベースから取得します。
今回のシミュレーションでは膜脂質を考慮しますが、配置予定の脂質分子がZ軸方向に沿うような系の座標になっている方が膜脂質を配置するソフトウェアにとって都合がよいです。
OPMに登録されている構造は、ソフトウェアの膜配置に適した座標/情報となっているため、今回はこちらから4ZJCの構造ファイルを取得します。
import os, requests
base_dir = "" # 任意のディレクトリの絶対パスを記載
# ファイルのダウンロード(4ZJ8: OX1)
pdb_id = "4ZJC"
pdb_id = pdb_id.lower()
url = f"https://biomembhub.org/shared/opm-assets/pdb/{pdb_id}.pdb"
os.makedirs(base_dir + "/data", exist_ok=True)
out_file = base_dir + f"/data/{pdb_id}_opm.pdb"
response = requests.get(url)
if response.status_code == 200:
with open(out_file, "wb") as f:
f.write(response.content)
else:
print(f"Failed to fetch {pdb_id} from OPM (status {response.status_code})")次に、biopythonを利用して、取得したPDBファイルからリガンドとタンパク質をそれぞれ別のファイルに分割します。
今回、シミュレーション時間の短縮のために、タンパク質は膜貫通領域でない部分を一部削除しました。
from Bio import PDB
# リガンドとレセプターの分割
protein_file = base_dir + f"/data/receptor_{pdb_id}.pdb"
ligand_file = base_dir + f"/data/ligand_{pdb_id}.pdb"
ligand_resname = "4OT"
parser = PDB.PDBParser(QUIET=True)
structure = parser.get_structure("complex", out_file)
io = PDB.PDBIO()
# レセプターを抽出
class ProteinSelect(PDB.Select):
def accept_residue(self, residue):
if not PDB.is_aa(residue, standard=True): # アミノ酸かどうか
return False
res_id = residue.id[1]
if 1001 <= res_id <= 1196: # 残基番号を取得 (id[1] が residue number)
return False # この範囲は除外
return True
io.set_structure(structure)
io.save(protein_file, ProteinSelect())
# リガンドを抽出
class LigandSelect(PDB.Select):
def accept_residue(self, residue):
return residue.get_resname().strip() == ligand_resname
io.set_structure(structure)
io.save(ligand_file, LigandSelect())次に、openbaelを利用して、リガンドのファイル形式を変換します(PDB→SDF)。
# リガンドのファイル形式を変換
ligand_sdf = base_dir + f"/data/ligand_{pdb_id}.sdf"
!obabel $ligand_file -O $ligand_sdf -p 7.4最後に、PDBFixerを利用して、タンパク質の構造をクレンジングします。
from pdbfixer import PDBFixer
from openmm.app import PDBFile
# PDBfixer
protein_file_fixed = base_dir + f"/data/receptor_{pdb_id}-fixed.pdb"
fixer = PDBFixer(filename=protein_file)
fixer.findNonstandardResidues()
fixer.replaceNonstandardResidues()
fixer.removeHeterogens(keepWater=False)
fixer.findMissingResidues()
fixer.findMissingAtoms()
fixer.addMissingAtoms()
fixer.addMissingHydrogens(pH=7.4)
with open(protein_file_fixed, "w") as f:
PDBFile.writeFile(fixer.topology, fixer.positions, f, keepIds=True)以上で、シミュレーションに利用するためのファイル準備は完了です。
系の準備
シミュレーションに必要となる情報や水分子やイオン・膜脂質を含んだ系を準備します。
利用する各種ライブラリをインポートしましょう。
import numpy as np
from rdkit import Chem
import mdtraj as md
import openmm as mm
import openmm.app as app
from openmm import unit
from openff.toolkit.topology import Molecule
from openmmforcefields.generators import GAFFTemplateGeneratorまずは、リガンドをOpenMMのオブジェクトに変換します。
# リガンドの前処理
ligname = "LIG"
supplier = Chem.SDMolSupplier(ligand_sdf, removeHs=False)
rdkit_mol = supplier[0]
# OpenFFのオブジェクトに変換
off_mol = Molecule.from_rdkit(rdkit_mol)
off_mol.name = ligname # リガンドの名前を指定
# 原子番号の指定
element_counter_dict = {}
for off_atom, rdkit_atom in zip(off_mol.atoms, rdkit_mol.GetAtoms()):
element = rdkit_atom.GetSymbol()
if element in element_counter_dict.keys():
element_counter_dict[element] += 1
else:
element_counter_dict[element] = 1
off_atom.name = element + str(element_counter_dict[element])
# OpenMMのオブジェクトに変換
off_mol_topology = off_mol.to_topology()
mol_topology = off_mol_topology.to_openmm()
mol_positions = off_mol.conformers[0]
mol_positions = mol_positions.to("nanometers") # 単位を変換:Å → nm
omm_mol = app.Modeller(mol_topology, mol_positions)続いて、リガンドを含むオブジェクトにタンパク質の情報を追加します。
# rガンド-タンパク質複合体の形成
md_protein_topology = md.Topology.from_openmm(fixer.topology) # PDBfixerのオブジェクトを利用
md_ligand_topology = md.Topology.from_openmm(omm_mol.topology)
md_complex_topology = md_protein_topology.join(md_ligand_topology)
complex_topology = md_complex_topology.to_openmm() # OpenMMのオブジェクトに変換
# 座標の更新
total_atoms = len(fixer.positions) + len(omm_mol.positions)
complex_positions = unit.Quantity(np.zeros([total_atoms, 3]), unit=unit.nanometers) # OpenMMで利用される配列
complex_positions[: len(fixer.positions)] = fixer.positions # タンパク質の座標を追加
complex_positions[len(fixer.positions) :] = omm_mol.positions # リガンドの座標を追加次に、力場の設定を行います。
今回は、タンパク質・脂質分子にamber14系、水分子にTIP3P-FB、リガンドにGAFF2を利用します。
# 力場の設定
protein_ff="amber14-all.xml" # タンパク質の力場(膜脂質の力場を含む)
solvent_ff="amber14/tip3pfb.xml" # 水の力場
forcefield = app.ForceField(protein_ff, solvent_ff)
gaff = GAFFTemplateGenerator(
molecules=Molecule.from_rdkit(rdkit_mol, allow_undefined_stereo=True) # リガンドの力場/電荷を設定
)
forcefield.registerTemplateGenerator(gaff.generator)リガンドとタンパク質のオブジェクトに水・イオン・膜脂質を追加します。また、完成したオブジェクトの情報をPDBファイルとして保存します。
# 複合体に水/イオン/膜脂質を追加
modeller = app.Modeller(complex_topology, complex_positions)
modeller.addMembrane(
forcefield,
lipidType="POPC",
membraneCenterZ=0.0*unit.nanometer, # 膜中心の Z 位置
minimumPadding=1.0*unit.nanometer, # タンパク質等との余裕 (10Å)
positiveIon="Na+",
negativeIon="Cl-",
ionicStrength=0.15*unit.molar
)
# PDBファイルとして保存
complex_file = base_dir + f"/data/complex_{pdb_id}.pdb"
with open(complex_file, "w") as f:
PDBFile.writeFile(modeller.topology, modeller.positions, f)以上で系の準備は完了です。
simulationオブジェクトの作成
シミュレーションに利用するオブジェクトを作成します。
バロスタットは膜脂質のシミュレーション向けに「MonteCarloMembraneBarostat」が実装されているのでそれを利用します。他の設定は膜なし場合と基本的に同じにしているつもりです。
# simulationオブジェクトを作成
# システム
system = forcefield.createSystem(
modeller.topology,
nonbondedMethod=app.PME,
nonbondedCutoff=1.0*unit.nanometer,
constraints='HBonds'
)
if not system.getDefaultPeriodicBoxVectors():
system.setDefaultPeriodicBoxVectors(*modeller.topology.getPeriodicBoxVectors())
# バロスタット
barostat = mm.MonteCarloMembraneBarostat(
1*unit.bar, # 圧力
0*unit.bar*unit.nanometer, # 表面張力(通常は0)
10*unit.kelvin, # 温度
mm.MonteCarloMembraneBarostat.XYIsotropic,
mm.MonteCarloMembraneBarostat.ZFree,
25 # 頻度
)
system.addForce(barostat)
# インテグレーター
integrator = mm.LangevinIntegrator(
10*unit.kelvin,
1/unit.picosecond,
0.002*unit.picoseconds
)
# プラットフォーム
platform = mm.Platform.getPlatformByName('CUDA') # または 'OpenCL', 'CPU'
properties = {'Precision': 'mixed'} # GPUの計算精度
# トポロジーと座標の取得
topology = modeller.getTopology()
positions = modeller.getPositions()
simulation = app.Simulation(topology, system, integrator, platform, properties)
simulation.context.setPositions(positions)MDシミュレーション
以下のステップでMDシミュレーションを実行します。
- エネルギー最適化
- 等温緩和
- 昇温
- 平衡化
- 生産
以下のコードで結果の保存先を作成しておきましょう。
result_dir = base_dir + "/result"
os.makedirs(result_dir, exist_ok=True)エネルギー最適化:Energy minimization (EM)
初速度を与える前に系のエネルギー最適化(EM)を実施します。
初期構造の歪を取り除くことによりポテンシャルを低下させ、最初期の破綻を防ぐ効果があります。
em_path = result_dir + '/1_minimization'
os.makedirs(em_path, exist_ok=True)
state_path_em = em_path + "/state.xml"
structure_path_em = em_path + "/structure.pdb"
# 実行
simulation.minimizeEnergy()
# 保存
state_em = simulation.context.getState(
getPositions=True,
getVelocities=True,
getEnergy=True,
getForces=True,
getParameters=True,
enforcePeriodicBox=True
)
with open(state_path_em, "w") as f:
f.write(mm.XmlSerializer.serialize(state_em))
positions_em = simulation.context.getState(getPositions=True).getPositions()
with open(structure_path_em, "w") as f:
PDBFile.writeFile(simulation.topology, positions_em, f)等温緩和:Quenched dynamics (QD)
初速度を与えてMDを開始します。
等温緩和(QD)は、10K以下の低温かつ定温でのMDを指します。EMと同様に、最初期に破綻を防ぐために実施します。
qd_path = result_dir + '/2_quenched_dynamics'
os.makedirs(qd_path, exist_ok=True)
state_path_qd = qd_path + "/state.xml"
structure_path_qd = qd_path + "/structure.pdb"
dcd_path_qd = qd_path + "/traj.dcd"
log_path_qd = qd_path + "/log.log"
# 設定
simulation.currentStep = 0
simulation.reporters = []
simulation.reporters.append(app.DCDReporter(dcd_path_qd, 2))
simulation.reporters.append(
app.StateDataReporter(
log_path_qd, 2, totalSteps=10, step=True, speed=True, progress=True, potentialEnergy=True, temperature=True
)
)
simulation.context.setState(state_em)
# 実行
simulation.step(10)
# 保存
state_qd = simulation.context.getState(
getPositions=True,
getVelocities=True,
getEnergy=True,
getForces=True,
getParameters=True,
enforcePeriodicBox=True
)
with open(state_path_qd, "w") as f:
f.write(mm.XmlSerializer.serialize(state_qd))
positions_qd = simulation.context.getState(getPositions=True).getPositions()
with open(structure_path_qd, "w") as f:
PDBFile.writeFile(simulation.topology, positions_qd, f)
昇温:heating
系の温度を段階的に上げていきます。今回は、以下の条件で昇温を実施しました。
- NPγTアンサンブル
- 拘束条件:膜脂質・タンパク質主鎖・リガンド、1,000 kJ/mol/nm2
- 温度:100K→300K(100K→150K→200K→250K→300K)
- 時間:0.5 ns(0.1ns→0.1ns→0.1ns→0.1ns→0.1ns)
ht_path = result_dir + '/3_heating'
os.makedirs(ht_path, exist_ok=True)
state_path_ht = ht_path + "/state.xml"
structure_path_ht = ht_path + "/structure.pdb"
dcd_path_ht = ht_path + "/traj.dcd"
log_path_ht = ht_path + "/log.log"
# 設定
n_steps = 250000 # 2fs * 250,000 = 500,000fs ← 500ps ← 0.5ns
simulation.context.setState(state_qd)
simulation.currentStep = 0
simulation.reporters = []
simulation.reporters.append(app.DCDReporter(dcd_path_ht, 5000))
simulation.reporters.append(
app.StateDataReporter(
log_path_ht, 5000, totalSteps=n_steps, step=True, speed=True, progress=True, potentialEnergy=True, temperature=True
)
)
# 拘束条件: 脂質、タンパク主鎖、リガンド
restraint = mm.CustomExternalForce("k*periodicdistance(x, y, z, x0, y0, z0)^2")
res_idx = system.addForce(restraint)
restraint.addGlobalParameter("k", 1000.0*unit.kilojoules_per_mole/unit.nanometer)
restraint.addPerParticleParameter("x0")
restraint.addPerParticleParameter("y0")
restraint.addPerParticleParameter("z0")
lipid_atoms = [atom for atom in topology.atoms() if atom.residue.name=="POP"]
for atom in lipid_atoms:
restraint.addParticle(atom.index, positions[atom.index])
protein_atoms = [atom for atom in topology.atoms() if atom.name in ["CA", "N", "C", "O"]]
for i in protein_atoms:
restraint.addParticle(atom.index, positions[atom.index])
ligand_atoms = [atom for atom in topology.atoms() if atom.residue.name=="LIG"]
for i in protein_atoms:
restraint.addParticle(atom.index, positions[atom.index])
# 実行
list_temp = [100, 150, 200, 250, 300]
list_step = [50000, 50000, 50000, 50000, 50000]
for temp, step in zip(list_temp, list_step):
integrator.setTemperature(temp*unit.kelvin)
barostat.setDefaultTemperature(temp*unit.kelvin)
simulation.step(step)
# 保存
state_ht = simulation.context.getState(
getPositions=True,
getVelocities=True,
getEnergy=True,
getForces=True,
getParameters=True,
enforcePeriodicBox=True
)
with open(state_path_ht, "w") as f:
f.write(mm.XmlSerializer.serialize(state_ht))
positions_ht = simulation.context.getState(getPositions=True).getPositions()
with open(structure_path_ht, "w") as f:
PDBFile.writeFile(simulation.topology, positions_ht, f)平衡化:equibaration
拘束条件を徐々に緩めるとともに、本計算に向けて系のエネルギーを平衡化します。今回は、以下の条件で平衡化を実施しました。
- NPγTアンサンブル
- 拘束条件:膜脂質・タンパク質主鎖・リガンド、1,000 kJ/mol/nm2→0 kJ/mol/nm2(1,000→300→100→30→0)
- 温度:300K
- 時間:0.5 ns(0.01ns→0.833..ns→0.833..ns→0.833..ns→0.25ns)
eq_path = result_dir + '/4_equibaration'
os.makedirs(eq_path, exist_ok=True)
state_path_eq = eq_path + "/state.xml"
structure_path_eq = eq_path + "/structure.pdb"
dcd_path_eq = eq_path + "/traj.dcd"
log_path_eq = eq_path + "/log.log"
# 設定
n_steps = 250000 # 2fs * 250,000 = 500,000fs ← 500ps ← 0.5ns
simulation.context.setState(state_ht)
simulation.currentStep = 0
simulation.reporters = []
simulation.reporters.append(app.DCDReporter(dcd_path_eq, 5000))
simulation.reporters.append(
app.StateDataReporter(
log_path_eq, 5000, totalSteps=n_steps, step=True, speed=True, progress=True, potentialEnergy=True, temperature=True
)
)
# 実行
list_restrain = [1000.0, 300.0, 100.0, 30.0, 0.0]
list_step = [5000, 40000, 40000, 40000, 125000]
for k, step in zip(list_restrain, list_step):
restraint.setGlobalParameterDefaultValue(0, k*unit.kilojoules_per_mole/unit.nanometer)
simulation.step(step)
# 保存
state_eq = simulation.context.getState(
getPositions=True,
getVelocities=True,
getEnergy=True,
getForces=True,
getParameters=True,
enforcePeriodicBox=True
)
with open(state_path_eq, "w") as f:
f.write(mm.XmlSerializer.serialize(state_eq))
positions_eq = simulation.context.getState(getPositions=True).getPositions()
with open(structure_path_eq, "w") as f:
PDBFile.writeFile(simulation.topology, positions_eq, f)本計算:Production run
最後に本計算を実施します。条件は以下の通りです。
- NPγTアンサンブル
- 拘束条件:なし
- 温度:300K
- 時間:1.0 ns
md_path = result_dir + '/5_production'
os.makedirs(md_path, exist_ok=True)
state_path_md = md_path + "/state.xml"
structure_path_md = md_path + "/structure.pdb"
dcd_path_md = md_path + "/traj.dcd"
log_path_md = md_path + "/log.log"
checkpoint_path = md_path + "/checkpoint.chk"
# 設定
n_steps = 500000 # 2fs * 500,000 = 1,000,000fs ← 1000ps ← 1.0ns
simulation.context.setState(state_eq)
simulation.currentStep = 0
simulation.reporters = []
simulation.reporters.append(app.DCDReporter(dcd_path_md, 5000))
simulation.reporters.append(
app.StateDataReporter(
log_path_md, 5000, totalSteps=n_steps, step=True, speed=True, progress=True, potentialEnergy=True, temperature=True
)
)
# 実行
simulation.step(n_steps)
# 保存
state_md = simulation.context.getState(
getPositions=True,
getVelocities=True,
getEnergy=True,
getForces=True,
getParameters=True,
enforcePeriodicBox=True
)
with open(state_path_md, "w") as f:
f.write(mm.XmlSerializer.serialize(state_md))
positions_md = simulation.context.getState(getPositions=True).getPositions()
with open(structure_path_md, "w") as f:
PDBFile.writeFile(simulation.topology, positions_md, f)
with open(checkpoint_path, "wb") as f:
f.write(simulation.context.createCheckpoint())
トラジェクトリの後処理
MDの結果を可視化する際、(ソフトウェアにもよりますが)未処理のファイルを読み込ませると分子がおかしな状態で表示されることがあります。
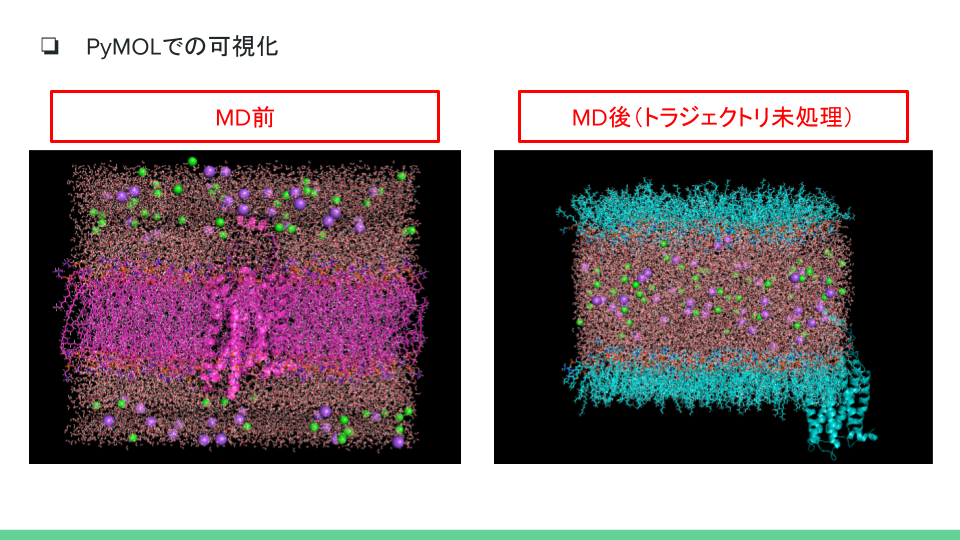
こちらを回避するためにトラジェクトリの後処理を行います。
fixed_path_ht = eq_path + "/traj_pc.dcd"
fixed_path_eq = ht_path + "/traj_pc.dcd"
fixed_path_md = md_path + "/traj_pc.dcd"
dict_path_dcd = {
dcd_path_ht:fixed_path_ht,
dcd_path_eq:fixed_path_eq,
dcd_path_md:fixed_path_md,
}
for dcd_path, fixed_dcd_path in dict_path_dcd.items():
traj = md.load(dcd_path, top=structure_path_em)
traj_whole = traj.make_molecules_whole() # 分子を “ちぎれない” 形に直す(make whole)
prot = traj_whole.topology.select("protein") # タンパクを基準にセンタリング&ボックス内にラップ
traj_centered = traj_whole.center_coordinates()
traj_wrapped = traj_centered.image_molecules() # 近接イメージに押し戻す
traj_fit = traj_wrapped.superpose(traj_wrapped, frame=0, atom_indices=prot) # 剛体合わせ(平行移動・回転除去)してRMSD等が滑らかに
traj_fit.save_dcd(fixed_dcd_path)後処理後のトラジェクトリを用いてMDを可視化すると、次のようになります(上:全体、下:リガンド周辺)。
本計算が1 nsということもありますが、実験構造が取得できるようなリガンドなのでかなり安定して結合して見えます。
最後に
「OpenMM」を用いた膜タンパク質+リガンドのMDシミュレーションを行いました。
途中、OpenMMの仕様にハマって挫けそうになりましたが、なんとかまとめることができてよかったです。この調子でFEPの計算もできるようになりたいですね。
以下、今回の検証で使用したコードです。本記事では紹介しませんでしたが、結果の解析も実施しています(MM/GBSA、MM/PBSA、LIE、RMSD、PCA、CC)。参考になれば幸いです。


コメント