

AQME (Automated Quantum Mechanical Environments) を使用して物性予測モデル構築用の特徴量を生成します。
実行環境
- OS: Ubuntu 22.04.3 LTS
- CPU: AMD Ryzen 7 5700X 8-Core Processor
- GPU: NVIDIA GeForce RTX 3060 Ti (8G)
- メモリ: 32GB
AQME について
AQMEは、量子化学(QM)計算のための各種ワークフローを提供する研究/教育用のOSSです。
分子のコンフォメーション探索や記述子の生成、QM計算ソフトウェアに応じたインプットファイル作成/後処理など、QM計算を円滑化するための各種機能が備わっています。
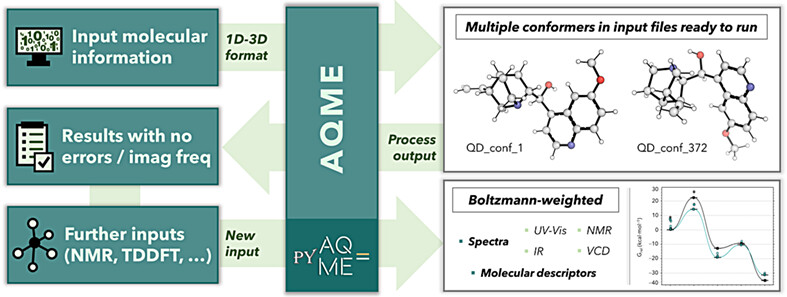
今回は、QM記述子を特徴量とした物性予測モデル構築のためにAQMEを使用したいと思います。
環境構築
ドキュメントの情報を参考にconda環境を構築します。
コンフォメーション探索や記述子の計算には、CRESTやxTBといった量子化学計算向けのライブラリを使用します。こちらはAQMEの依存パッケージに含まれていないので別途インストールする必要があります。
また、コンフォメーションの最適化段階には、RDkitの力場やxTBに加えて機械学習ポテンシャルを利用することができます。今回は使いませんが、今後の利用を考えて使えるようにしておきましょう。
以下、私の環境で実際に利用したコマンドです。
conda create -n aqme python=3.10
conda activate aqme
pip install aqme
conda install -y -c conda-forge openbabel=3.1.1
conda install -y -c conda-forge xtb=6.7.1
conda install -y -c conda-forge crest=2.12
conda install conda-forge::libgfortran=14.2.0
pip install ase
pip install torch==2.5.1 torchvision==0.20.1 torchaudio==2.5.1 --index-url https://download.pytorch.org/whl/cu121
pip install torchani今回は、最後に予測モデルを用いた検証も行います。
AQMEには必要ありませんが、以下のパッケージをインストールしておくとコードが再現できるので、必要に応じてインストールしましょう。
pip install scikit-learn
pip install matplotlib
pip install lightgbm
pip install seabornAQME の操作
Githubに公開されているnotebookを参考にQM記述子を計算します。
計算対象のデータセットは、MolecularNetの「Lipophilicity(LogD)」です。LogDの予測にAQMEで算出されたQM記述子がどの程度有用か確認してみましょう。
※以下の操作は、新規のディレクトリ内に作成したnotebookファイルのjupyterでの実行を想定しています。
まずは、DeepChemが提供しているLipophilicityのデータセットをダウンロードします。
import os
import pandas as pd
base_dir = '' # 新規作成したディレクトリの絶対パス
# データの取得
df = pd.read_csv('https://deepchemdata.s3-us-west-1.amazonaws.com/datasets/Lipophilicity.csv')
df.rename(columns={'smiles':'SMILES', 'CMPD_CHEMBLID':'code_name'}, inplace=True)
os.makedirs(base_dir + '/dataset', exist_ok=True)
df.to_csv(base_dir + '/dataset/lipophilicity.csv', index=False)次に、Lipophilicityに含まれる化合物のコンフォメーション探索を実施します。
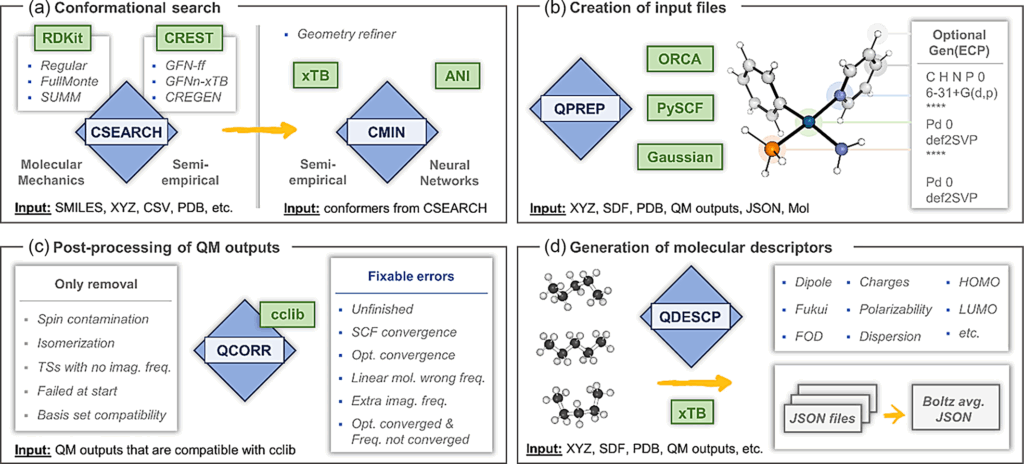
AQMEの「CSEARCH」は、化合物の構造ファイルを入力として、コンフォメーションの生成〜最適化までを実施します。
バックグラウンドのプログラムとしては、RDkitとCRESTが選択可能です。今回はRDkitを利用したコンフォメーション生成と力場レベルでのコンフォメーション最適化までを実施します(時間がかかります。私の環境では16時間弱でした)。
from aqme.csearch import csearch
csearch(
input='dataset/lipophilicity.csv',
program='rdkit',
w_dir_main=base_dir,
sample=25, # コンフォメーションの生成数
ff='MMFF', # 最適化に利用する力場
ewin_csearch=5, # エネルギーの閾値
nprocs=12 # プロセス数(要検討)
)次に、生成したコンフォメーションを利用してQM記述子を計算します。
AQMEの「QDESCP」では、半経験的量子化学計算の手法であるxTB (extended Tight Binding) を用いた計算を行い、各化合物について入力コンフォメーションのボルツマン分布を考慮したQM計算値(ボルツマン重み付け平均)を出力します(半経験的なので正確にはSQM記述子?)。
ソースコードを見る限りですが、xTBの計算は真空条件による最適化と溶媒モデル(ALPB model)を利用したエネルギー計算の双方を行っているようです(間違っていたらすみません)。
実行は以下のコードです(時間がかかります。私の環境では約3日でした)。
from aqme.qdescp import qdescp
sdf_rdkit_files = glob.glob(f'CSEARCH/*.sdf')
qdescp(
program='xtb',
files=sdf_rdkit_files,
robert=False, # ROBERT用のデータベース作成
nprocs=12, # プロセス数(要検討)
boltz=True, # ボルツマン重み付け平均+RDkit記述子の算出
qdescp_temp=300, # QM計算に使用する温度
)上記のコードで各化合物のQM記述子が記載されたcsvファイルが作成されますが、今回使用したデータではボルツマン重み付け平均の段階で計算エラーが発生しストップしました
ただし、エラーまでにQM記述子の算出に必要なデータはできていたので、エラー回避可能なコードを作成し望みのファイルを作成し続きの処理を行いました(後述。もとのコードをChatGPTに与えて作成)。
以上で、QM記述の計算は完了です。ちなみに、本来出力されるのcsvファイルはQM記述子と一緒にRDkit記述子も含まれていますが、ChatGPTに書かせたコードではRDkit記述子は含まれていませんでした。もともとQM記述子とRDkit記述子は分ける予定だったので結果オーライです。
予測モデル構築
①QM記述子、②RDkit記述子、③QM記述子+RDkit記述子、この3パターンの特徴量で予測モデルを作成し、LogD予測に対するQM記述子の有用性を考察したいと思います。
今回は、データセットを訓練データとテストデータにランダム分割(8対2)し、LightGBMのデフォルト設定でモデルを構築して予測精度を確認します。
まずは必要なライブラリをインポートしましょう。
import matplotlib.pyplot as plt
from lightgbm import LGBMRegressor
from rdkit import Chem
from rdkit.Chem import Descriptors
from rdkit import RDLogger
RDLogger.DisableLog('rdApp.*')
import seaborn as sns
import pandas as pd
from sklearn.metrics import root_mean_squared_error, r2_score次に、Lipophilicityのデータと特徴量のデータを統合します。
# AQMEの特徴量をデータセットと紐付ける
df_base = pd.read_csv(base_dir + '/dataset/lipophilicity.csv')
df_desc = pd.read_csv(qdescp_dir + '/QDESCP_full_descriptors.csv')
df_desc['code_name'] = df_desc['code_name'].map(lambda x: x.split('_')[0]) # _rdkitの消去
df_aqme = pd.merge(df_base, df_desc, on='code_name', how='left')
df_aqme.to_csv(base_dir + '/dataset/lipophilicity_aqme.csv', header=True, index=False)
# RDkit descriptorをデータセットと紐付ける
desc_json = {}
for name, smi in zip(df_base['code_name'], df_base['SMILES']):
mol = Chem.MolFromSmiles(smi)
descrs = Descriptors.CalcMolDescriptors(mol)
desc_json[name] = descrs
df_rddesc = pd.DataFrame(data=desc_json).T.reset_index(names='code_name')
df_rddesc = pd.merge(df_base, df_rddesc, on='code_name', how='left')
df_rddesc.to_csv(base_dir + '/dataset/lipophilicity_rddesc.csv', header=True, index=False)各パターンの特徴量を用いた予測モデルを構築します。
# 予測モデル構築:AQME
df_aqme_test = df_aqme.sample(frac=0.2, random_state=0)
df_aqme_train = df_aqme.drop(index=df_aqme_test.index)
y_test, X_aqme_test = df_aqme_test['exp'], df_aqme_test.drop(columns=['code_name', 'exp', 'SMILES'])
y_train, X_aqme_train = df_aqme_train['exp'], df_aqme_train.drop(columns=['code_name', 'exp', 'SMILES'])
model_aqme = LGBMRegressor(random_state=42, verbosity=0)
model_aqme.fit(X_aqme_train, y_train)
# 予測モデル構築:RDkit descriptor
df_rddesc_test = df_rddesc.sample(frac=0.2, random_state=0)
df_rddesc_train = df_rddesc.drop(index=df_rddesc_test.index)
X_rddesc_test = df_rddesc_test.drop(columns=['code_name', 'exp', 'SMILES'])
X_rddesc_train = df_rddesc_train.drop(columns=['code_name', 'exp', 'SMILES'])
model_rddesc = LGBMRegressor(random_state=42, verbosity=0)
model_rddesc.fit(X_rddesc_train, y_train)
# 予測モデル構築:AQME + RDkit descriptor
X_mix_test = pd.concat([X_aqme_test, X_rddesc_test], axis=1)
X_mix_train = pd.concat([X_aqme_train, X_rddesc_train], axis=1)
model_mix = LGBMRegressor(random_state=42, verbosity=0)
model_mix.fit(X_mix_train, y_train)テストデータを予測して各モデルの精度を確認しましょう。
# 評価
y_pred_test_aqme = model_aqme.predict(X_aqme_test)
y_pred_test_rddesc = model_rddesc.predict(X_rddesc_test)
y_pred_test_mix = model_mix.predict(X_mix_test)
rmsd_aqme = root_mean_squared_error(y_test, y_pred_test_aqme)
rmsd_rddesc = root_mean_squared_error(y_test, y_pred_test_rddesc)
rmsd_mix = root_mean_squared_error(y_test, y_pred_test_mix)
r2_aqme = r2_score(y_test, y_pred_test_aqme)
r2_rddesc = r2_score(y_test, y_pred_test_rddesc)
r2_mix = r2_score(y_test, y_pred_test_mix)
print(f'model AQME: RMSE={rmsd_aqme:.3f}', f'R2={r2_aqme:.3f}')
print(f'model RDkit descriptor: RMSE={rmsd_rddesc:.3f}', f'R2={r2_rddesc:.3f}')
print(f'model AQME+RDkit descripotr: RMSE={rmsd_mix:.3f}', f'R2={r2_mix:.3f}')
# プロット
fig, ax = plt.subplots(1, 3, figsize=(18, 6))
ax[0].scatter(y_test, y_pred_test_aqme, label=f'R2: {r2_aqme:.3f}')
ax[0].set_title('AQME')
ax[0].set_xlabel('ture val')
ax[0].set_ylabel('pred val')
ax[0].legend()
ax[1].scatter(y_test, y_pred_test_rddesc, label=f'R2: {r2_rddesc:.3f}')
ax[1].set_title('RDkit descriptor')
ax[1].set_xlabel('ture val')
ax[1].set_ylabel('pred val')
ax[1].legend()
ax[2].scatter(y_test, y_pred_test_mix, label=f'R2: {r2_mix:.3f}')
ax[2].set_title('AQME+RDkit descriptor')
ax[2].set_xlabel('ture val')
ax[2].set_ylabel('pred val')
ax[2].legend()
plt.show()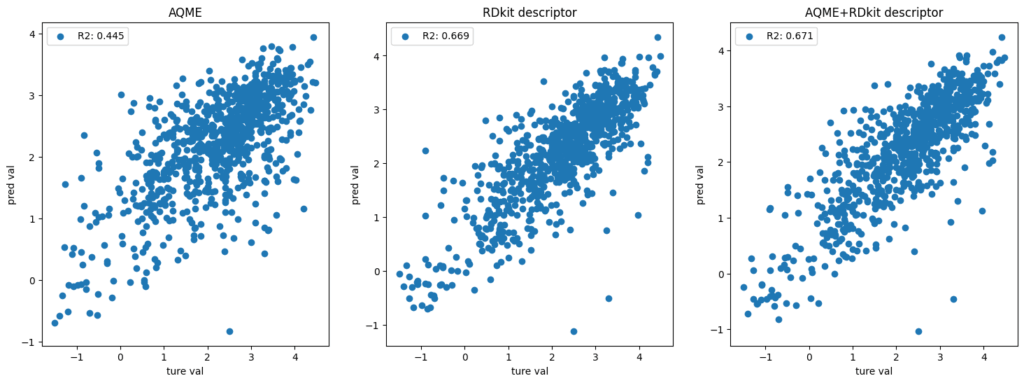
今回の検証は条件設定がかなり荒いものの、RDkit記述子を用いたモデルの精度が最も高く、QM記述子単独ではそこまで精度が出ないという結果となりました。
また、特徴量を組み合わせたモデルでは決定係数の上昇が認められていますが、本当に意味がある上昇かは微妙です。
最後に、各モデルの特徴量の寄与度を可視化してみます。
# 寄与度の可視化
cont_aqme = pd.Series(model_aqme.feature_importances_, index=model_aqme.feature_names_in_).sort_values(ascending=False).iloc[:10]
cont_rddesc = pd.Series(model_rddesc.feature_importances_, index=model_rddesc.feature_names_in_).sort_values(ascending=False).iloc[:10]
cont_mix = pd.Series(model_mix.feature_importances_, index=model_mix.feature_names_in_).sort_values(ascending=False).iloc[:10]
fig, ax = plt.subplots(1, 3, figsize=(30, 12))
cont_aqme[::-1].plot(ax=ax[0], kind='barh')
ax[0].set_title('AQME')
ax[0].set_xlabel('importance')
cont_rddesc[::-1].plot(ax=ax[1], kind='barh')
ax[1].set_title('RDkit descriptor')
ax[1].set_xlabel('importance')
cont_mix[::-1].plot(ax=ax[2], kind='barh')
ax[2].set_title('AQME+RDkit descriptor')
ax[2].set_xlabel('importance')
plt.show()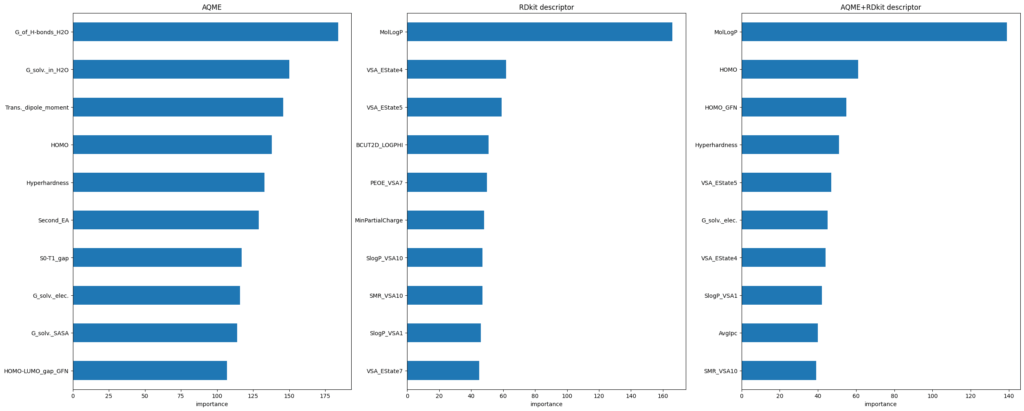
QM記述子のモデルでは水の溶媒効果や双極子モーメントが寄与度の上位に来ており、原理的に考えても一定の納得感があります。RDkit記述子もMolLogPが上位に来るのは妥当ですね。
特徴量を組み合わせたモデルでは、2番目以降にQM記述子がそこそこ入ってきています。今回はLightGBMのデフォルト設定でモデルを構築したためこのような結果となりましたが、特徴量選択やハイパーパラメータ調整でもう少し違った見え方となるかもしれません。
最後に
「AQME」を用いたQM記述子の算出とLogD予測を行いました。
分子の3D記述子は一部の物性やADMET予測において、0~2Dの記述子を補完する形で有用性が報告されているため、今回の結果も妥当なのではないかと思います。
QMの計算結果はケミストの理解を得やすい印象があるため、いざというときに結果を利用できるような環境を整えたいですね。
以下、今回の検証で使用したコードです。参考になれば幸いです。


コメント