

オープンソースソフトウェアを活用したドッキングシミュレーション及び結合自由エネルギー評価を実施します。
実行環境
- OS: Ubuntu 22.04.3 LTS
- CPU: AMD Ryzen 7 5700X 8-Core Processor
- GPU: NVIDIA GeForce RTX 3060 Ti (8G)
- RAM: 32GB
概要
大規模化合物ライブラリから有望な候補を抽出するバーチャルスクリーニングやAI創薬では、標的タンパク質の立体構造情報を活用した Structure-Based Drug Design(SBDD)の手法が中核を担っています。
ドッキングシミュレーションは、標的に対する結合ポーズを静的に推定し、結合親和性を簡易的にスコアリングすることで、候補化合物群を効率的に濃縮する探索的手段として広く利用されています。
濃縮された化合物群はさらに、自由エネルギー摂動法(FEP)や分子力学的自由エネルギー法(MM/GBSA, MM/PBSA)によって再スコアリングされ、より定量的な評価を経てヒット率の向上が図られます。
一般的なSBDDによるスコアリングフローは、次の4段階から構成されます。
- 標的構造および対象化合物の準備(大前提)
- リガンドの三次元構造生成
- ドッキングシミュレーション
- 結合自由エネルギー評価
今回は、リガンド三次元化に「Gypsum-DL」、ドッキングに「GNINA」、自由エネルギー評価に「Uni-GBSA」を用いることで、オープンソースツールのみで再現可能なSBDDスコアリングパイプラインを構築します。
環境構築
Gypsum-DL、Uni-MM/GBSAの環境構築にはcondaを利用し、GNINAの環境構築にはDockerを利用します。
conda環境
以下の手順で環境を構築します。2025年10月現在、Gypsum-DLはpipのインストールのみではコマンドが利用できないため、リポジトリをローカル環境にクローンして.pyファイルをたたく必要があります。
conda create -n gbsa -c conda-forge acpype openmpi mpi4py gromacs "gmx_mmpbsa>=1.5.6"
conda activate gbsa
pip install unigbsa lickit
conda install pdbfixer biopython
pip install gypsum-dl
cd (任意のディレクトリ)
git clone https://github.com/durrantlab/gypsum_dl.git
cd gypsum_dl
git checkout v1.2.1Docker環境
公式が配布しているイメージファイルを利用してGNINAの環境を構築します。以下のコマンドで導入できますが、ファイル容量が大きい(45G)ので注意しましょう。
docker pull gnina/gnina※nvidia-docker2の導入が必要です。導入に関しては、以下の記事が参考になれば幸いです。

パイプライン構築
以下、jupyter notebook上での作業想定でパイプライン構築を行います。
標的構造および対象化合物の準備
対象化合物として、ChEMBLに登録されているEGFRのリガンドを利用します。
以前に”ChEMBL websource client”で取得したデータから20化合物を抜粋して利用します。

コードは以下の通りです。
import os
from rdkit.Chem import PandasTools
base_dir = "" # .ipynbが存在するディレクトリを指定
data_src_path = "" # 以前に取得したEGFRデータ (.sdf) のパスを指定
data_dist_sdf_path = base_dir + "/data/EGFR.sdf"
data_dist_smi_path = base_dir + "/data/EGFR.smi"
gypsum_output_dir = base_dir + "/data/3dmol"
# データを抽出
df = PandasTools.LoadSDF(data_src_path)
df_select = df.sample(n=20, random_state=42)
# .smiとして保存:Gypsum-DLで利用
df_smi = df_select.loc[:, ["smiles", "ID"]]
df_smi.to_csv(data_dist_smi_path, index=False, header=False, sep="\t")
# .sdfとして保存:GNINAで利用
for name, mol in zip(df["ID"], df["ROMol"]):
mol.SetProp("_Name", name)
df_select.drop(columns="ID", inplace=True)
PandasTools.WriteSDF(df_select, data_dist_sdf_path, properties=list(df_select.columns))次に、標的タンパク質の構造を処理します。今回は、PDBに登録されている「2ITO」を利用します。
ドッキングシミュレーションでは、シミュレーションボックス(結合ポーズ推定時の探索範囲)を指定する必要があります。探索の中心座標を入力する必要がありますが、GNINAでは参照リガンドのSDFを与えることで自動で中心を設定してくれます。
複合体の構造が既知の場合は、参照リガンドの準備も行いましょう。
import requests
from pdbfixer import PDBFixer
from openmm.app import PDBFile
from Bio import PDB
pdb_id = "2ITO"
url = f"https://files.rcsb.org/download/{pdb_id}.pdb"
out_file = base_dir + f"/data/{pdb_id}.pdb"
ligand_pdb_file = base_dir + f"/data/ligand_{pdb_id}.pdb"
ligand_sdf_file = base_dir + f"/data/ligand_{pdb_id}.sdf"
protein_file = base_dir + f"/data/receptor_{pdb_id}.pdb"
# ファイルのダウンロード
response = requests.get(url)
if response.status_code == 200:
with open(out_file, "wb") as f:
f.write(response.content)
else:
print(f"Failed to fetch {pdb_id}")
# リガンドを抽出:GNINAで利用
ligand_resname = "IRE"
parser = PDB.PDBParser(QUIET=True)
structure = parser.get_structure("complex", out_file)
io = PDB.PDBIO()
# リガンドを抽出
class LigandSelect(PDB.Select):
def accept_residue(self, residue):
return residue.get_resname().strip() == ligand_resname
io.set_structure(structure)
io.save(ligand_pdb_file, LigandSelect())
# リガンドのファイル形式を変換
!obabel $ligand_pdb_file -O $ligand_sdf_file -p 7.4
# クレンジング
fixer = PDBFixer(filename=out_file)
fixer.findNonstandardResidues()
fixer.replaceNonstandardResidues()
fixer.removeHeterogens(keepWater=False)
fixer.findMissingResidues()
fixer.findMissingAtoms()
fixer.addMissingAtoms()
fixer.addMissingHydrogens(pH=7.4)
with open(protein_file, "w") as f:
PDBFile.writeFile(fixer.topology, fixer.positions, f, keepIds=True)リガンドの三次元構造生成
Gypsum-DLは、SBDDの前処理において「リガンドの三次元構造を生成・整備する」ための強力なオープンソースツールです。
二次元構造をインプットとすることで、三次元構造や立体異性体、プロトン化状態を自動で生成します。
ドッキングシミュレーションでは、生体内の環境を考慮したリガンドの三次元化が重要になりますが、Gypsum-DLは大量の化合物を高速かつ正確に処理することが可能です。
個人的な印象としては、やはり有償ソフトのwash機能と比べると精度は下がります。一方で、大規模な化合物ライブラリを対象としたケースでは十分に有用だと感じています。
さて、リガンドの三次元化に関しては、以下のコードで実行します。引数の意味についてはGithubを参考にしていただけますと幸いです。
# リガンドの3次元化
%cd (ローカル環境にクローンしたGypsum-DLのディレクトリを指定)
!python run_gypsum_dl.py \
--source $data_dist_smi_path \
--output_folder $gypsum_output_dir \
--job_manager multiprocessing \
--num_processors 8 \
--max_variants_per_compound 1 \
--separate_output_files \
--min_ph 7.4 \
--max_ph 7.4 \
--skip_making_tautomers \
--skip_enumerate_chiral_mol \
--skip_enumerate_double_bonds
%cd $base_dirドッキングシミュレーション
GNINAは、3D-Convolutional Neural Network (CNN) を統合したドッキングシミュレーションソフトウェアです。
オープンソースのドッキングソフトウェアである AutoDock Vina をベースとしており、配座のスコアリング部分に対して、リガンド-タンパク質の複合体構造を3D-CNNで学習したAIモデルが利用されています。
GNINAを利用したドッキングシミュレーションは、以下のコードで実行します。GNINAのコンテナ作成〜破棄までを連続で行っているので、利用の際は注意しましょう。
from rdkit import Chem
import pandas as pd
gnina_dir = base_dir + "/gnina"
os.makedirs(gnina_dir, exist_ok=True)
docked_dir = base_dir + "/docked"
os.makedirs(docked_dir, exist_ok=True)
docked_top_dir = docked_dir + "/top"
os.makedirs(docked_top_dir, exist_ok=True)
# コンテナを立ち上げる
!docker run -d \
--name gnina \
-v $base_dir:/root/base \
--gpus all \
gnina/gnina \
tail -f /dev/null
# ドッキングシミュレーション
list_sdf = [f for f in os.listdir(gypsum_output_dir) if not "gypsum" in f]
for sdf in list_sdf:
!docker exec -it gnina \
gnina \
-r /root/base/data/receptor_2ITO.pdb \
-l /root/base/data/3dmol/{sdf} \
--autobox_ligand /root/base/data/ligand_2ITO.sdf \
--exhaustiveness 32 \
-o /root/base/gnina/{sdf}.gz
!docker exec -it gnina \
gunzip /root/base/gnina/{sdf}.gz
# コンテナの削除
!docker container stop gnina
!docker container rm gninaGNINAで生成されたドッキングポーズはプロトンの情報が不足しています。この状態ではUni-GBSAの計算時に都合が悪いため、以下のコードで再プロトン化します。
# プロトン化
for sdf in list_sdf:
!obabel $gnina_dir/{sdf} -O $docked_dir/{sdf} -p 7.4また、生成されたドッキングポーズのSDFには、GNINAのスコアリングされた上位~10化合物が保存されています。Uni-GBSA及び解析のために最上位のポーズのみを取得して保存しましょう。
# スコア最上位の分割
df_gnina = pd.DataFrame()
for sdf in list_sdf:
_df = PandasTools.LoadSDF(docked_dir + f"/{sdf}", removeHs=False)
_df = _df.loc[0, :]
df_gnina = pd.concat([df_gnina, _df], axis=1)
_df["ROMol"].SetProp("_Name", _df["ID"])
_df["ROMol"].SetProp("SMILES", _df["SMILES"])
_df["ROMol"].SetProp("Energy", _df["Energy"])
_df["ROMol"].SetProp("minimizedAffinity", _df["minimizedAffinity"])
_df["ROMol"].SetProp("CNNscore", _df["CNNscore"])
_df["ROMol"].SetProp("CNNaffinity", _df["CNNaffinity"])
_df["ROMol"].SetProp("CNN_VS", _df["CNN_VS"])
_df["ROMol"].SetProp("CNNaffinity_variance", _df["CNNaffinity_variance"])
writer = Chem.SDWriter(docked_top_dir + f"/{sdf}")
writer.write(_df["ROMol"])
writer.close()
df_gnina.T.to_csv(docked_top_dir + "/gnina_result.scv", index=False)結合自由エネルギー評価
Uni-GBSAは、分子力学+溶媒モデル(MM/GBSA 、MM/PBSA)で結合自由エネルギーを高速に推定するためのオープンソースツールであり、MDシミュレーションを必要とせずドッキングポーズのみを利用することが特徴です。
期待する精度としては、ドッキングシミュレーションのような経験則のスコアリング以上、FEP及び複数スナップショットを用いたMM/GB (PB) SA未満、といったところです。
Uni-GBSAを用いた結合自由エネルギー計算は、以下のコードで実行します。
引数の詳細はGithubに記載されている通りですが、2025年10月現在、一部の引数が機能していないような挙動が認められています(例えば、出力ファイルの名前を決める -o が反映されない)。
また、引数に絶対パスが指定できないようなので使い方には注意が必要です。
# Uni-GBSA
%cd !base_dir
for sdf in list_sdf:
gbsa_dir = base_dir + f"/uni_gbsa/{sdf.split('.')[0]}"
os.makedirs(gbsa_dir, exist_ok=True)
%cd $gbsa_dir
!unigbsa-pipeline \
-i ../../data/receptor_2ITO.pdb \
-l ../../docked/top/{sdf} \
-o BindingEnergy.csv
%cd !base_dir最後に、解析用に結果をcsvファイルに統合します。
# データの集約
df_gbsa = pd.DataFrame()
for sdf in list_sdf:
gbsa_path = base_dir + f"/uni_gbsa/{sdf.split('.')[0]}/BindingEnergy.csv"
_df = pd.read_csv(gbsa_path)
df_gbsa = pd.concat([df_gbsa, _df], axis=0)
df_gbsa.to_csv(base_dir + "/uni_gbsa/gbsa_result.csv", index=False)
df_gbsa実測値 vs 計算値
最後に、実測のpIC50とドッキングスコア(mimimizedAffinity、CNNaffinity)及び結合自由エネルギー(MM/GBSA)をプロットしてどの程度相関が認められるか確認してみます。
import matplotlib.pyplot as plt
# データの読み込み
df_base = PandasTools.LoadSDF(data_dist_sdf_path)
df_gnina = pd.read_csv(docked_top_dir + "/gnina_result.scv")
df_gbsa = pd.read_csv(base_dir + "/uni_gbsa/gbsa_result.csv")
# ID順に並べ替え
df_base.sort_values(by="ID", ascending=True, inplace=True)
df_gnina.sort_values(by="ID", ascending=True, inplace=True)
df_gbsa.sort_values(by="ligandName", ascending=True, inplace=True)
# マージ
df_base = pd.concat(
[
df_base,
df_gnina.loc[:, ["minimizedAffinity", "CNNaffinity"]],
df_gbsa.loc[:, "TOTAL"]
],
axis=1
)
df_base
# float化
df_base["pIC50"] = df_base["pIC50"].astype(float)
df_base["minimizedAffinity"] = df_base["minimizedAffinity"].astype(float)
df_base["CNNaffinity"] = df_base["CNNaffinity"].astype(float)
df_base["TOTAL"] = df_base["TOTAL"].astype(float)
# プロット
r2_smina = df_base["pIC50"].corr(df_base["minimizedAffinity"])**2
r2_gnina = df_base["pIC50"].corr(df_base["CNNaffinity"])**2
r2_gbsa = df_base["pIC50"].corr(df_base["TOTAL"])**2
fig, ax = plt.subplots(1, 3, figsize=(18, 6))
ax[0].scatter(df_base["pIC50"], df_base["minimizedAffinity"], c="r", alpha=0.5, label=f"R2:{r2_smina:.3}")
ax[0].invert_yaxis()
ax[0].set_xlabel("pIC50")
ax[0].set_ylabel("minimizedAffinity")
ax[0].legend()
ax[1].scatter(df_base["pIC50"], df_base["CNNaffinity"], c="g", alpha=0.5, label=f"R2:{r2_gnina:.3}")
ax[1].set_xlabel("pIC50")
ax[1].set_ylabel("CNNaffinity")
ax[1].legend()
ax[2].scatter(df_base["pIC50"], df_base["TOTAL"], c="b", alpha=0.5, label=f"R2:{r2_gbsa:.3}")
ax[2].invert_yaxis()
ax[2].set_xlabel("pIC50")
ax[2].set_ylabel("⊿G MM/GBSA")
ax[2].legend()
plt.show()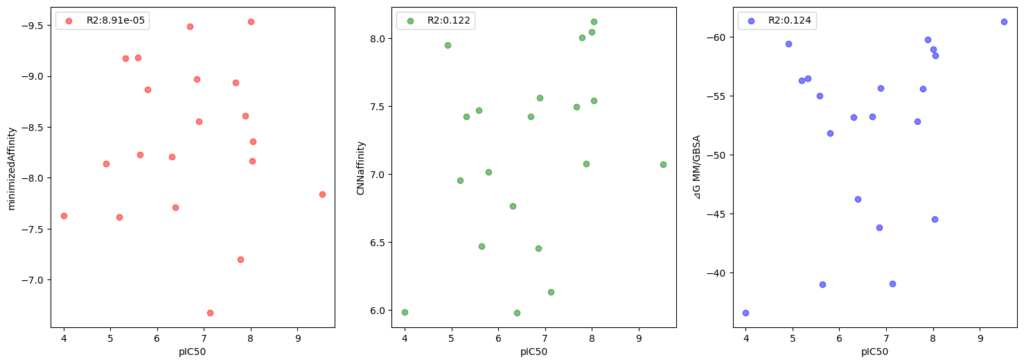
左から、minimizedAffinity(sminaのスコア)、CNNaffinity(GNINAのスコア)、MM/GBSAの⊿Gとなります。
全体的な相関は良いとは言えませんが、sminaのスコアよりもGNINAのスコア、GNINAのスコアよりもMM/GBSAの⊿G、といったような精度の傾向が見て取れます。このデータだけをみると、ヒット率を上げるためにはMM/GBSAまで計算してスコアリングするのが良さそうに思えます。
実際には、標的タンパク質や利用する複合体構造、評価するリガンドの骨格、計算のパラメータ等、実測値との相関に与える影響は多岐に渡ります。また、計算量が上がるからと言って必ずしも精度向上につながるわけではありません。
どのスコアリングを利用するかはケースバイケースで対応を変える必要があることを念頭に置きたいですね。
最後に
オープンソースソフトウェアを活用したドッキングシミュレーション及び結合自由エネルギー評価を実施しました。
当初の予定としては、Uni-GBSAだけを検討する予定でしたが、せっかくなので実務に近いフローを考えて実装してみました(カスタマイズ次第で実務応用も可能だと思います)。
また、今回利用したコード(.ipynb)はこちらから利用可能です。参考になれば幸いです

コメント